Circrnas in Host And viRuses anaLysis pIpEline
![]()
![]()





See the website for detailed information, documentation, and examples: https://ccbr.github.io/CHARLIE/
Circrnas in Host And viRuses anaLysis pIpEline
Things to know about CHARLIE:
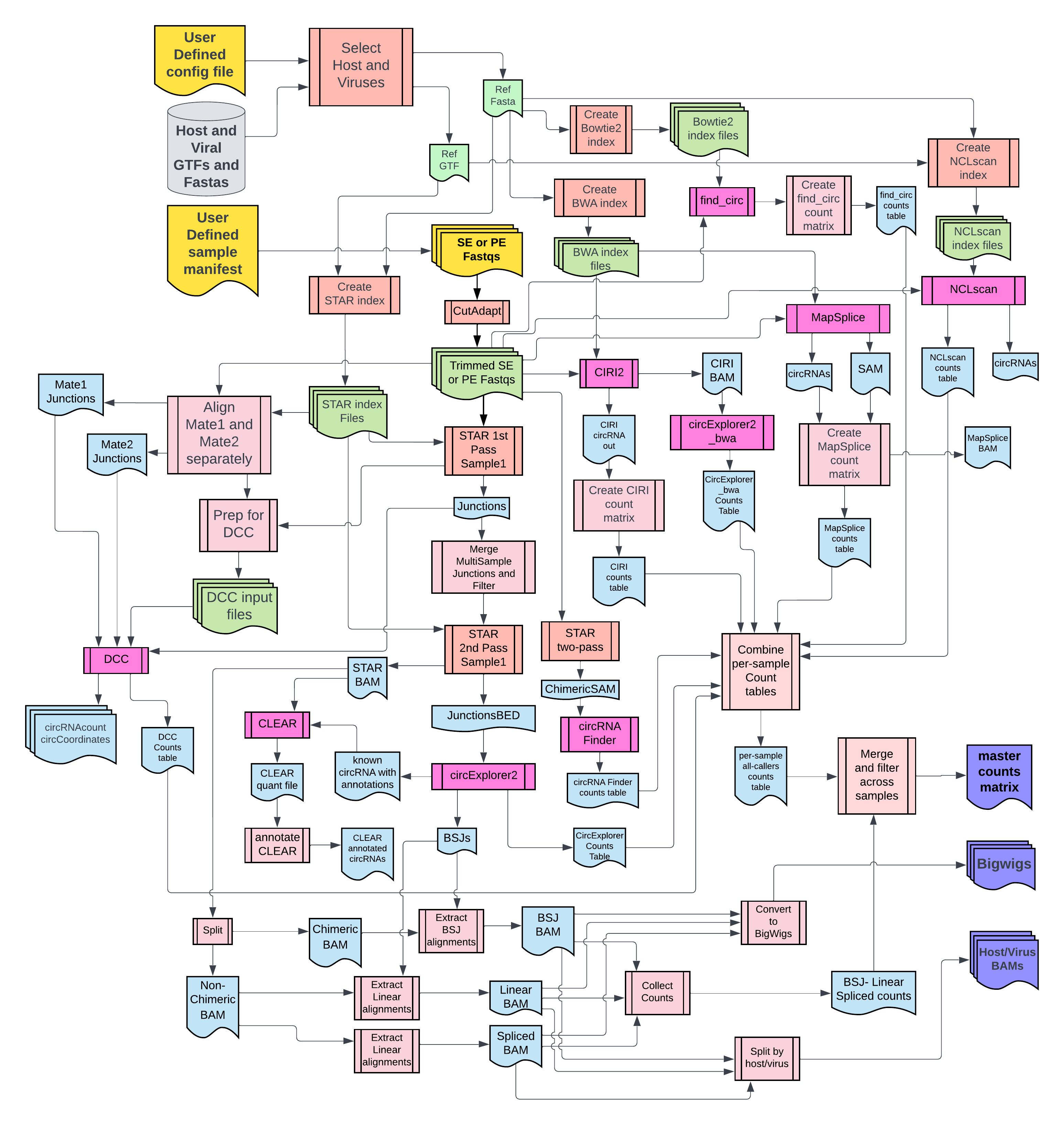
- Snakemake workflow to detect, annotate and quantify (DAQ) host and viral circular RNAs.
- Primirarily developed to run on BIOWULF
- Reach out to Vishal Koparde for questions/comments/requests.
This circularRNA detection pipeline uses CIRCExplorer2, CIRI2 and many other tools in parallel to detect, quantify and annotate circRNAs. Here is a list of tools that can be run using CHARLIE:
| circRNA Detection Tool | Aligner(s) | Run by default |
|---|---|---|
| CIRCExplorer2 | STAR1 | Yes |
| CIRI2 | BWA1 | Yes |
| CIRCExplorer2 | BWA1 | Yes |
| CLEAR | STAR1 | Yes |
| DCC | STAR2 | Yes |
| circRNAFinder | STAR3 | Yes |
| find_circ | Bowtie2 | Yes |
| MapSplice | BWA2 | No |
| NCLScan | NovoAlign | No |
Note: STAR1, STAR2, STAR3 denote 3 different sets of alignment parameters, etc.
Note: BWA1, BWA2 denote 2 different alignment parameters, etc.
For complete documentation, view the website https://CCBR.github.io/CHARLIE/.
⚠️ DISCLAIMER: New circRNA tools have been added CHARLIE and the documentation is currently out of date!
The following version of various bioinformatics tools are using within CHARLIE:
| tool | version |
|---|---|
| blat | 3.5 |
| bedtools | 2.30.0 |
| bowtie | 2-2.5.1 |
| bowtie | 1.3.1 |
| bwa | 0.7.17 |
| circexplorer2 | 2.3.8 |
| cufflinks | 2.2.1 |
| cutadapt | 4.4 |
| fastqc | 0.11.9 |
| hisat | 2.2.2.1 |
| java | 18.0.1.1 |
| multiqc | 1.9 |
| parallel | 20231122 |
| perl | 5.34 |
| picard | 2.27.3 |
| python | 2.7 |
| python | 3.8 |
| sambamba | 0.8.2 |
| samtools | 1.16.1 |
| STAR | 2.7.6a |
| stringtie | 2.2.1 |
| ucsc | 450 |
| R | 4.0.5 |
| novocraft | 4.03.05 |
% ./charlie
##########################################################################################
Welcome to
_______ __ __ _______ ______ ___ ___ _______
| || | | || _ || _ | | | | | | |
| || |_| || |_| || | || | | | | | ___|
| || || || |_||_ | | | | | |___
| _|| || || __ || |___ | | | ___|
| |_ | _ || _ || | | || || | | |___
|_______||__| |__||__| |__||___| |_||_______||___| |_______|
C_ircrnas in H_ost A_nd vi_R_uses ana_L_ysis p_I_p_E_line
##########################################################################################
This pipeline was built by CCBR (https://bioinformatics.ccr.cancer.gov/ccbr)
Please contact Vishal Koparde for comments/questions ([email protected])
##########################################################################################
CHARLIE can be used to DAQ(Detect/Annotate/Quantify) circRNAs in hosts and viruses.
Here is the list of hosts and viruses that are currently supported:
HOSTS:
* hg38 [Human]
* mm39 [Mouse]
ADDITIVES:
* ERCC [External RNA Control Consortium sequences]
* BAC16Insert [insert from rKSHV.219-derived BAC clone of the full-length KSHV genome]
VIRUSES:
* NC_007605.1 [Human gammaherpesvirus 4 (Epstein-Barr virus)]
* NC_006273.2 [Human betaherpesvirus 5 (Cytomegalovirus )]
* NC_001664.4 [Human betaherpesvirus 6A (HHV-6A)]
* NC_000898.1 [Human betaherpesvirus 6B (HHV-6B)]
* NC_001716.2 [Human betaherpesvirus 7 (HHV-7)]
* NC_009333.1 [Human gammaherpesvirus 8 (KSHV)]
* NC_045512.2 [Severe acute respiratory syndrome(SARS)-related coronavirus]
* MN485971.1 [HIV from Belgium]
* NC_001806.2 [Human alphaherpesvirus 1 (Herpes simplex virus type 1)](strain 17) (HSV-1)]
* KT899744.1 [HSV-1 strain KOS]
* MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]
##########################################################################################
USAGE:
bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>
Required Arguments:
1. WORKDIR : [Type: String]: Absolute or relative path to the output folder with write permissions.
2. RUNMODE : [Type: String] Valid options:
* init : initialize workdir
* dryrun : dry run snakemake to generate DAG
* run : run with slurm
* runlocal : run without submitting to sbatch
ADVANCED RUNMODES (use with caution!!)
* unlock : unlock WORKDIR if locked by snakemake NEVER UNLOCK WORKDIR WHERE PIPELINE IS CURRENTLY RUNNING!
* reconfig : recreate config file in WORKDIR (debugging option) EDITS TO config.yaml WILL BE LOST!
* reset : DELETE workdir dir and re-init it (debugging option) EDITS TO ALL FILES IN WORKDIR WILL BE LOST!
* printbinds: print singularity binds (paths)
* local : same as runlocal
Optional Arguments:
--singcache|-c : singularity cache directory. Default is `/data/${USER}/.singularity` if available, or falls back to `${WORKDIR}/.singularity`. Use this flag to specify a different singularity cache directory.
--host|-g : supply host at command line. hg38 or mm39. (--runmode=init only)
--additives|-a : supply comma-separated list of additives at command line. ERCC or BAC16Insert or both (--runmode=init only)
--viruses|-v : supply comma-separated list of viruses at command line (--runmode=init only)
--manifest|-s : absolute path to samples.tsv. This will be copied to output folder (--runmode=init only)
--changegrp|-z : change group to "Ziegelbauer_lab" before running anything. Biowulf-only. Useful for correctly setting permissions.
--help|-h : print this help
Example commands:
bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=init
bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=dryrun
bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=run
##########################################################################################
VersionInfo:
python : 3.7
snakemake : 7.19.1
pipeline_home : /vf/users/Ziegelbauer_lab/Pipelines/circRNA/activeDev
git commit/tag : 1ae5ca091976364369784f67adffbbbf1dcdb7d5 v0.8-197-g1ae5ca0
##########################################################################################MIT License
Copyright (c) 2021 Vishal Koparde
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
Run init mode:
bash <path to charlie> -w=<path to output dir> -m=initThis will create the folder provided by -w=. The user should have write permission to this folder.
Test data (1 paired-end subsample and 1 single-end subsample) have been including under the .tests/dummy_fastqs folder. After running in -m=init, samples.tsv should be edited to point the copies of the above mentioned samples with the column headers:
- sampleName
- path_to_R1_fastq
- path_to_R2_fastq
Column path_to_R2_fastq will be blank in case of single-end samples.
After editing samples.tsv, dry run should be run:
bash <path to charlie> -w=<path to output dir> -m=dryrunThis will create the reference fasta and gtf file based on the selections made in the config.yaml.
If -m=dryrun was successful, then simply do -m=run. The output will look something like this
... ... skipping ~1000 lines
...
...
Job stats:
job count min threads max threads
--------------------------------------------- ------- ----------
all 1 1 1
annotate_clear_output 2 1 1
circExplorer 2 2 2
circExplorer_bwa 2 2 2
circrnafinder 2 1 1
ciri 2 56 56
clear 2 2 2
create_bowtie2_index 1 1 1
create_bwa_index 1 1 1
create_circExplorer_BSJ_bam 2 4 4
create_circExplorer_linear_spliced_bams 2 56 56
create_circExplorer_merged_found_counts_table 2 1 1
create_hq_bams 2 1 1
create_index 1 56 56
create_master_counts_file 1 1 1
cutadapt 2 56 56
dcc 2 4 4
dcc_create_samplesheets 2 1 1
estimate_duplication 2 1 1
fastqc 2 4 4
find_circ 2 56 56
find_circ_align 2 56 56
merge_SJ_tabs 1 2 2
merge_alignment_stats 1 1 1
merge_genecounts 1 1 1
merge_per_sample 2 1 1
star1p 2 56 56
star2p 2 56 56
star_circrnafinder 2 56 56
total 52 1 56
Reasons:
(check individual jobs above for details)
input files updated by another job:
alignment_stats, all, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_master_counts_file, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder
missing output files:
alignment_stats, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_bowtie2_index, create_bwa_index, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_index, create_master_counts_file, cutadapt, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
Running...
14743440
The .tests/dummy_fastqs folder in the repo has test dataset:
% tree .tests/dummy_fastqs
.tests/dummy_fastqs
├── GI1_N.R1.fastq.gz
├── GI1_N.R2.fastq.gz
└── GI1_T.R1.fastq.gzGI1_N is a PE sample while GI1_T is a SE sample.
Expected output from the sample data is stored under .tests/expected_output.
More details about running test data can be found here.
DISCLAIMER:
CHARLIE is built to be run only on BIOWULF. A newer HPC-agnostic version of CHARLIE is planned for 2024.