\ No newline at end of file
+ XAVIER Documentation
404 - Not found
\ No newline at end of file
diff --git a/dev/contributing/index.html b/dev/contributing/index.html
new file mode 100644
index 0000000..8b1c71c
--- /dev/null
+++ b/dev/contributing/index.html
@@ -0,0 +1,18 @@
+ How to contribute - XAVIER Documentation
If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
Create a Git branch for your pull request (PR). Give the branch a descriptive name for the changes you will make, such as iss-10 if it is for a specific issue.
# create a new branch and switch to it
+gitbranchiss-10
+gitswitchiss-10
+
Changes to the python package code will also need unit tests to demonstrate that the changes work as intended. We write unit tests with pytest and store them in the tests/ subdirectory. Run the tests with python -m pytest.
If you change the workflow, please run the workflow with the test profile and make sure your new feature or bug fix works as intended.
If you're not sure how often you should commit or what your commits should consist of, we recommend following the "atomic commits" principle where each commit contains one new feature, fix, or task. Learn more about atomic commits here: https://www.freshconsulting.com/insights/blog/atomic-commits/
First, add the files that you changed to the staging area:
gitaddpath/to/changed/files/
+
Then make the commit. Your commit message should follow the Conventional Commits specification. Briefly, each commit should start with one of the approved types such as feat, fix, docs, etc. followed by a description of the commit. Take a look at the Conventional Commits specification for more detailed information about how to write commit messages.
gitcommit-m'feat: create function for awesome feature'
+
pre-commit will enforce that your commit message and the code changes are styled correctly and will attempt to make corrections if needed.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Failed
hook id: trailing-whitespace
exit code: 1
files were modified by this hook > Fixing path/to/changed/files/file.txt > codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped
In the example above, one of the hooks modified a file in the proposed commit, so the pre-commit check failed. You can run git diff to see the changes that pre-commit made and git status to see which files were modified. To proceed with the commit, re-add the modified file(s) and re-run the commit command:
gitaddpath/to/changed/files/file.txt
+gitcommit-m'feat: create function for awesome feature'
+
This time, all the hooks either passed or were skipped (e.g. hooks that only run on R code will not run if no R files were committed). When the pre-commit check is successful, the usual commit success message will appear after the pre-commit messages showing that the commit was created.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Passed codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped Conventional Commit......................................................Passed > [iss-10 9ff256e] feat: create function for awesome feature 1 file changed, 22 insertions(+), 3 deletions(-)
Finally, push your changes to GitHub:
gitpush
+
If this is the first time you are pushing this branch, you may have to explicitly set the upstream branch:
gitpush--set-upstreamoriginiss-10
+
Enumerating objects: 7, done. Counting objects: 100% (7/7), done. Delta compression using up to 10 threads Compressing objects: 100% (4/4), done. Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done. Total 4 (delta 3), reused 0 (delta 0), pack-reused 0 remote: Resolving deltas: 100% (3/3), completed with 3 local objects. remote: remote: Create a pull request for 'iss-10' on GitHub by visiting: remote: https://github.com/CCBR/XAVIER/pull/new/iss-10 remote: To https://github.com/CCBR/XAVIER > > [new branch] iss-10 -> iss-10 branch 'iss-10' set up to track 'origin/iss-10'.
We recommend pushing your commits often so they will be backed up on GitHub. You can view the files in your branch on GitHub at https://github.com/CCBR/XAVIER/tree/<your-branch-name> (replace <your-branch-name> with the actual name of your branch).
Edit the PR title and description. The title should briefly describe the change. Follow the comments in the template to fill out the body of the PR, and you can delete the comments (everything between <!-- and -->) as you go. Be sure to fill out the checklist, checking off items as you complete them or striking through any irrelevant items. When you're ready, click 'Create pull request' to open it.
Optionally, you can mark the PR as a draft if you're not yet ready for it to be reviewed, then change it later when you're ready.
We will do our best to follow the tidyverse code review principles: https://code-review.tidyverse.org/. The reviewer may suggest that you make changes before accepting your PR in order to improve the code quality or style. If that's the case, continue to make changes in your branch and push them to GitHub, and they will appear in the PR.
Once the PR is approved, the maintainer will merge it and the issue(s) the PR links will close automatically. Congratulations and thank you for your contribution!
\ No newline at end of file
diff --git a/dev/faq/questions/index.html b/dev/faq/questions/index.html
index 65752c6..44f7267 100644
--- a/dev/faq/questions/index.html
+++ b/dev/faq/questions/index.html
@@ -1 +1 @@
- General Questions - XAVIER Documentation
\ No newline at end of file
diff --git a/dev/index.html b/dev/index.html
index a929b96..f5f7b94 100644
--- a/dev/index.html
+++ b/dev/index.html
@@ -1,4 +1,4 @@
- XAVIER Documentation
_XAVIER - eXome Analysis and Variant explorER_. This is the home of the pipeline, XAVIER. Its long-term goals: to accurately call germline and somatic variants, to infer CNVs, and to boldly annotate variants like no pipeline before!
Welcome to XAVIER! Before getting started, we highly recommend reading through xavier's documentation.
The xavier pipeline is composed several inter-related sub commands to setup and run the pipeline across different systems. Each of the available sub commands perform different functions:
xavier run: Run the XAVIER pipeline with your input files.
xavier unlock: Unlocks a previous runs output directory.
XAVIER is a comprehensive whole exome-sequencing pipeline following the Broad's set of best practices. It relies on technologies like Singularity1 to maintain the highest-level of reproducibility. The pipeline consists of a series of data processing and quality-control steps orchestrated by Snakemake2, a flexible and scalable workflow management system, to submit jobs to a cluster or cloud provider.
The pipeline is compatible with data generated from Illumina short-read sequencing technologies. As input, it accepts a set of FastQ or BAM files and can be run locally on a compute instance, on-premise using a cluster, or on the cloud (feature coming soon!). A user can define the method or mode of execution. The pipeline can submit jobs to a cluster using a job scheduler like SLURM, or run on AWS using Tibanna (feature coming soon!). A hybrid approach ensures the pipeline is accessible to all users.
Before getting started, we highly recommend reading through the usage section of each available sub command.
For more information about issues or trouble-shooting a problem, please checkout our FAQ prior to opening an issue on Github.
Snakemake and singularity must be installed on the target system. Snakemake orchestrates the execution of each step in the pipeline. To guarantee the highest level of reproducibility, each step relies on versioned images from DockerHub. Snakemake uses singaularity to pull these images onto the local filesystem prior to job execution, and as so, snakemake and singularity are the only two dependencies.
_XAVIER - eXome Analysis and Variant explorER_. This is the home of the pipeline, XAVIER. Its long-term goals: to accurately call germline and somatic variants, to infer CNVs, and to boldly annotate variants like no pipeline before!
Welcome to XAVIER! Before getting started, we highly recommend reading through xavier's documentation.
The xavier pipeline is composed several inter-related sub commands to setup and run the pipeline across different systems. Each of the available sub commands perform different functions:
xavier run: Run the XAVIER pipeline with your input files.
xavier unlock: Unlocks a previous runs output directory.
XAVIER is a comprehensive whole exome-sequencing pipeline following the Broad's set of best practices. It relies on technologies like Singularity1 to maintain the highest-level of reproducibility. The pipeline consists of a series of data processing and quality-control steps orchestrated by Snakemake2, a flexible and scalable workflow management system, to submit jobs to a cluster or cloud provider.
The pipeline is compatible with data generated from Illumina short-read sequencing technologies. As input, it accepts a set of FastQ or BAM files and can be run locally on a compute instance, on-premise using a cluster, or on the cloud (feature coming soon!). A user can define the method or mode of execution. The pipeline can submit jobs to a cluster using a job scheduler like SLURM, or run on AWS using Tibanna (feature coming soon!). A hybrid approach ensures the pipeline is accessible to all users.
Before getting started, we highly recommend reading through the usage section of each available sub command.
For more information about issues or trouble-shooting a problem, please checkout our FAQ prior to opening an issue on Github.
Snakemake and singularity must be installed on the target system. Snakemake orchestrates the execution of each step in the pipeline. To guarantee the highest level of reproducibility, each step relies on versioned images from DockerHub. Snakemake uses singaularity to pull these images onto the local filesystem prior to job execution, and as so, snakemake and singularity are the only two dependencies.
# XAVIER is configured to use different execution backends: local or slurm# view the help page for more information
moduleloadccbrpipeliner
xavierrun--help
diff --git a/dev/license/index.html b/dev/license/index.html
index 46b53d8..8378bdb 100644
--- a/dev/license/index.html
+++ b/dev/license/index.html
@@ -1 +1 @@
- License - XAVIER Documentation
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
\ No newline at end of file
+ License - XAVIER Documentation
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
\ No newline at end of file
diff --git a/dev/pipeline-details/methods/index.html b/dev/pipeline-details/methods/index.html
index 9f27bf1..2be811c 100644
--- a/dev/pipeline-details/methods/index.html
+++ b/dev/pipeline-details/methods/index.html
@@ -1 +1 @@
- Methods - XAVIER Documentation
Low-quality and adapters sequences are trimmed from the raw sequencing reads using Trimmomatic (v. 0.39)1. Trimmed reads are then aligned to the human hg38 reference genome using BWA mapping software (v. 0.7.17)2. Duplicate reads are marked using Samblaster (v. 0.1.25)3 and sorted using samtools (v. 1.8). Finally, base quality score recalibration is performed as indicated in the GATK4 (v. 4.2.2.0) best practices 4.
HaplotypeCaller from GATK4 (v. 4.2.2.0) is used to call germline variants, parallelized across chromosomes, and all samples in the cohort are joint genotyped together 4,5.
Somatic variant calling (SNPs and Indels) is performed using Mutect (v. 1.1.7)6, Mutect2 (GATK v. 4.2.0)7, Strelka2 (v. 2.9.0)8, and VarDict (v. 1.4)9 in tumor-normal mode. Variants from all callers are merged using the CombineVariants tool from GATK version 3.8-1. Genomic, functional and consequence annotations are added using Variant Effect Predictor (VEP v. 99)10 and converted to Mutation Annotation Format (MAF) using the vcf2maf tool (v. 1.6.16)11.
For Copy Number Variants (CNVs), Control-Freec (v. 11.6)12 is used to generate pileups, which are used as input for the R package 'sequenza' (v. 3.0.0)13. The complete Control-Freec workflow is then re-run using ploidy and cellularity estimates from 'sequenza'.
SOBDetector is a tool that scores variants based on strand-orientation bias, which can be a sign of DNA damage caused by fixation of tissue. This pipeline runs SOBDetector in a two-pass method. The first pass uses parameters provided with the software (calculated from publicly available data from TCGA), then cohort-specific bias metrics are computed from those results, and SOBDetector is re-run using these cohort-specific values.
Ancestry and relatedness scores are generated using Somalier (v. 0.2.13)14. Contamination analyses are performed against viral and bacterial genomes from NCBI using Kraken2 (v. 2.1.2)15, as well as against mouse, human, and UniVec databases using FastQ Screen (v. 0.14.1)16. Sequence, mapping and variant statistics are computed using FastQC (v. 0.11.9), Qualimap (v. 2.2.1)17 and SNPeff (v. 4.3t)18. All of these metrics are combined into an interactive HTML report using MultiQC (v. 1.11)19.
Bolger, A.M., M. Lohse, and B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114-20. ↩
Li, H. and R. Durbin, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009. 25(14): p. 1754-60. ↩
Faust, G.G. and I.M. Hall, SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics, 2014. 30(17): p. 2503-5. ↩
Van der Auwera, G.A. and B.D. O'Connor, Genomics in the cloud : using Docker, GATK, and WDL in Terra. First edition. ed. 2020, Sebastopol, CA: O'Reilly Media. ↩↩
Poplin, R., et al., Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv, 2018: p. 201178. ↩
Cibulskis, K., et al., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol, 2013. 31(3): p. 213-9. ↩
Benjamin, D., et al., Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 2019: p. 861054. ↩
Kim, S., et al., Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods, 2018. 15(8): p. 591-594. ↩
Lai, Z., et al., VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res, 2016. 44(11): p. e108. ↩
McLaren, W., et al., The Ensembl Variant Effect Predictor. Genome Biol, 2016. 17(1): p. 122. ↩
Boeva, V., et al., Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics, 2012. 28(3): p. 423-5. ↩
Favero, F., et al., Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol, 2015. 26(1): p. 64-70. ↩
Pedersen, B. somalier: extract informative sites, evaluate relatedness, and perform quality-control on BAM/CRAM/BCF/VCF/GVCF. 2018; Available from: https://github.com/brentp/somalier. ↩
Wood, D.E., J. Lu, and B. Langmead, Improved metagenomic analysis with Kraken 2. Genome Biol, 2019. 20(1): p. 257. ↩
Wingett, S.W. and S. Andrews, FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res, 2018. 7: p. 1338. ↩
Okonechnikov, K., A. Conesa, and F. Garcia-Alcalde, Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics, 2016. 32(2): p. 292-4. ↩
Cingolani, P., et al., A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 2012. 6(2): p. 80-92. ↩
Ewels, P., et al., MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 2016. 32(19): p. 3047-8. ↩
Koster, J. and S. Rahmann, Snakemake-a scalable bioinformatics workflow engine. Bioinformatics, 2018. 34(20): p. 3600. ↩
\ No newline at end of file
+ Methods - XAVIER Documentation
Low-quality and adapters sequences are trimmed from the raw sequencing reads using Trimmomatic (v. 0.39)1. Trimmed reads are then aligned to the human hg38 reference genome using BWA mapping software (v. 0.7.17)2. Duplicate reads are marked using Samblaster (v. 0.1.25)3 and sorted using samtools (v. 1.8). Finally, base quality score recalibration is performed as indicated in the GATK4 (v. 4.2.2.0) best practices 4.
HaplotypeCaller from GATK4 (v. 4.2.2.0) is used to call germline variants, parallelized across chromosomes, and all samples in the cohort are joint genotyped together 4,5.
Somatic variant calling (SNPs and Indels) is performed using Mutect (v. 1.1.7)6, Mutect2 (GATK v. 4.2.0)7, Strelka2 (v. 2.9.0)8, and VarDict (v. 1.4)9 in tumor-normal mode. Variants from all callers are merged using the CombineVariants tool from GATK version 3.8-1. Genomic, functional and consequence annotations are added using Variant Effect Predictor (VEP v. 99)10 and converted to Mutation Annotation Format (MAF) using the vcf2maf tool (v. 1.6.16)11.
For Copy Number Variants (CNVs), Control-Freec (v. 11.6)12 is used to generate pileups, which are used as input for the R package 'sequenza' (v. 3.0.0)13. The complete Control-Freec workflow is then re-run using ploidy and cellularity estimates from 'sequenza'.
SOBDetector is a tool that scores variants based on strand-orientation bias, which can be a sign of DNA damage caused by fixation of tissue. This pipeline runs SOBDetector in a two-pass method. The first pass uses parameters provided with the software (calculated from publicly available data from TCGA), then cohort-specific bias metrics are computed from those results, and SOBDetector is re-run using these cohort-specific values.
Ancestry and relatedness scores are generated using Somalier (v. 0.2.13)14. Contamination analyses are performed against viral and bacterial genomes from NCBI using Kraken2 (v. 2.1.2)15, as well as against mouse, human, and UniVec databases using FastQ Screen (v. 0.14.1)16. Sequence, mapping and variant statistics are computed using FastQC (v. 0.11.9), Qualimap (v. 2.2.1)17 and SNPeff (v. 4.3t)18. All of these metrics are combined into an interactive HTML report using MultiQC (v. 1.11)19.
Bolger, A.M., M. Lohse, and B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114-20. ↩
Li, H. and R. Durbin, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009. 25(14): p. 1754-60. ↩
Faust, G.G. and I.M. Hall, SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics, 2014. 30(17): p. 2503-5. ↩
Van der Auwera, G.A. and B.D. O'Connor, Genomics in the cloud : using Docker, GATK, and WDL in Terra. First edition. ed. 2020, Sebastopol, CA: O'Reilly Media. ↩↩
Poplin, R., et al., Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv, 2018: p. 201178. ↩
Cibulskis, K., et al., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol, 2013. 31(3): p. 213-9. ↩
Benjamin, D., et al., Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 2019: p. 861054. ↩
Kim, S., et al., Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods, 2018. 15(8): p. 591-594. ↩
Lai, Z., et al., VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res, 2016. 44(11): p. e108. ↩
McLaren, W., et al., The Ensembl Variant Effect Predictor. Genome Biol, 2016. 17(1): p. 122. ↩
Boeva, V., et al., Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics, 2012. 28(3): p. 423-5. ↩
Favero, F., et al., Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol, 2015. 26(1): p. 64-70. ↩
Pedersen, B. somalier: extract informative sites, evaluate relatedness, and perform quality-control on BAM/CRAM/BCF/VCF/GVCF. 2018; Available from: https://github.com/brentp/somalier. ↩
Wood, D.E., J. Lu, and B. Langmead, Improved metagenomic analysis with Kraken 2. Genome Biol, 2019. 20(1): p. 257. ↩
Wingett, S.W. and S. Andrews, FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res, 2018. 7: p. 1338. ↩
Okonechnikov, K., A. Conesa, and F. Garcia-Alcalde, Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics, 2016. 32(2): p. 292-4. ↩
Cingolani, P., et al., A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 2012. 6(2): p. 80-92. ↩
Ewels, P., et al., MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 2016. 32(19): p. 3047-8. ↩
Koster, J. and S. Rahmann, Snakemake-a scalable bioinformatics workflow engine. Bioinformatics, 2018. 34(20): p. 3600. ↩
\ No newline at end of file
diff --git a/dev/pipeline-details/output/index.html b/dev/pipeline-details/output/index.html
index 72738ac..93fcf38 100644
--- a/dev/pipeline-details/output/index.html
+++ b/dev/pipeline-details/output/index.html
@@ -1,4 +1,4 @@
- Output Files - XAVIER Documentation
The output files and their locations are broken down here for the XAVIER pipeline. Pre-processing and germline variant calling steps are common but somatic variant calling is dependent on whether the pipeline was run in either (A) tumor-normal pair or (B) tumor-only analysis mode. All file locations are relative to the output directory specified during the job submission.
The output directory after a complete XAVIER run should look like:
The output files and their locations are broken down here for the XAVIER pipeline. Pre-processing and germline variant calling steps are common but somatic variant calling is dependent on whether the pipeline was run in either (A) tumor-normal pair or (B) tumor-only analysis mode. All file locations are relative to the output directory specified during the job submission.
The output directory after a complete XAVIER run should look like:
xavier_output/
├──bams
├──cluster.json# cluster info for the run
├──config
diff --git a/dev/pipeline-details/overview/index.html b/dev/pipeline-details/overview/index.html
index 56e16c3..f1801b6 100644
--- a/dev/pipeline-details/overview/index.html
+++ b/dev/pipeline-details/overview/index.html
@@ -1 +1 @@
- Overview - XAVIER Documentation
Workflow diagram of the XAVIER: the pipeline is composed of a series of data processing steps to trim, align, and recalibrate reads prior to calling variants. These data processing steps closely follow GATK's best pratices for cleaning up raw alignments. The pipeline also consists of a series of comprehensive quality-control steps.
\ No newline at end of file
+ Overview - XAVIER Documentation
Workflow diagram of the XAVIER: the pipeline is composed of a series of data processing steps to trim, align, and recalibrate reads prior to calling variants. These data processing steps closely follow GATK's best pratices for cleaning up raw alignments. The pipeline also consists of a series of comprehensive quality-control steps.
\ No newline at end of file
diff --git a/dev/pipeline-details/settings/index.html b/dev/pipeline-details/settings/index.html
index 94fc3d3..08710f1 100644
--- a/dev/pipeline-details/settings/index.html
+++ b/dev/pipeline-details/settings/index.html
@@ -1 +1 @@
- Settings - XAVIER Documentation
Removes variants with certain flags from vardict; (1) Germline: detected in germline sample (pass all quality parameters); (2) LikelyLOH: detected in germline but either lost in tumor OR 20-80% in germline, but increased to 1-opt_V (95%); (3) AFDiff: detected in tumor (pass quality parameters) and present in germline but didn’t pass quality parameters.
varscan
pileup
-d 100000 -q 15 -Q 15
samtools mpileup arguments; max depth of 100,000; min mapping quality of 15; min base quality of 15
calling
--strand-filter 0
Do not ignore variants with >90% support on one strand
--min-var-freq 0.01
Minimum variant allele frequency threshold 0.01
--output-vcf 1
Output in VCF format
--variants 1
Report only variant (SNP/indel) positions
all
GATK SelectVariants
--exclude-filtered
Removes non-PASS variants
--discordance
Remove variants found in supplied file (same as panel-of-normals file)
Removes variants with certain flags from vardict; (1) Germline: detected in germline sample (pass all quality parameters); (2) LikelyLOH: detected in germline but either lost in tumor OR 20-80% in germline, but increased to 1-opt_V (95%); (3) AFDiff: detected in tumor (pass quality parameters) and present in germline but didn’t pass quality parameters.
varscan
pileup
-d 100000 -q 15 -Q 15
samtools mpileup arguments; max depth of 100,000; min mapping quality of 15; min base quality of 15
calling
--strand-filter 0
Do not ignore variants with >90% support on one strand
--min-var-freq 0.01
Minimum variant allele frequency threshold 0.01
--output-vcf 1
Output in VCF format
--variants 1
Report only variant (SNP/indel) positions
all
GATK SelectVariants
--exclude-filtered
Removes non-PASS variants
--discordance
Remove variants found in supplied file (same as panel-of-normals file)
Make sure you're keeping the changelog up-to-date during development. Ideally, every PR that includes a user-facing change (e.g. a new feature, bug fix, or any API change) should add a concise summary to the changelog with a link to the PR. Only approve or merge PRs that either update the changelog or have no user-facing changes.
Edit the heading for the development version to match the new version.
If needed, clean up the changelog -- fix any typos, optionally create subheadings for 'New features' and 'Bug fixes' if there are lots of changes, etc.
Make sure you're keeping the changelog up-to-date during development. Ideally, every PR that includes a user-facing change (e.g. a new feature, bug fix, or any API change) should add a concise summary to the changelog with a link to the PR. Only approve or merge PRs that either update the changelog or have no user-facing changes.
Edit the heading for the development version to match the new version.
If needed, clean up the changelog -- fix any typos, optionally create subheadings for 'New features' and 'Bug fixes' if there are lots of changes, etc.
# go to the shared pipeline directory on biowulfcd/data/CCBR_Pipeliner/Pipelines/XAVIER
# clone the new version tag (e.g. v3.0.2) to a hidden directory
diff --git a/dev/search/search_index.json b/dev/search/search_index.json
index 448cc50..8c5ac67 100644
--- a/dev/search/search_index.json
+++ b/dev/search/search_index.json
@@ -1 +1 @@
-{"config":{"lang":["en"],"separator":"[\\s\\-]+","pipeline":["stopWordFilter"]},"docs":[{"location":"","title":"About","text":""},{"location":"#xavier-exome-analysis-and-variant-explorer","title":"XAVIER - eXome Analysis and Variant explorER \ud83d\udd2c","text":"
_XAVIER - eXome Analysis and Variant explorER_. This is the home of the pipeline, XAVIER. Its long-term goals: to accurately call germline and somatic variants, to infer CNVs, and to boldly annotate variants like no pipeline before!
Welcome to XAVIER! Before getting started, we highly recommend reading through xavier's documentation.
The xavier pipeline is composed several inter-related sub commands to setup and run the pipeline across different systems. Each of the available sub commands perform different functions:
xavier run: Run the XAVIER pipeline with your input files.
xavier unlock: Unlocks a previous runs output directory.
XAVIER is a comprehensive whole exome-sequencing pipeline following the Broad's set of best practices. It relies on technologies like Singularity1 to maintain the highest-level of reproducibility. The pipeline consists of a series of data processing and quality-control steps orchestrated by Snakemake2, a flexible and scalable workflow management system, to submit jobs to a cluster or cloud provider.
The pipeline is compatible with data generated from Illumina short-read sequencing technologies. As input, it accepts a set of FastQ or BAM files and can be run locally on a compute instance, on-premise using a cluster, or on the cloud (feature coming soon!). A user can define the method or mode of execution. The pipeline can submit jobs to a cluster using a job scheduler like SLURM, or run on AWS using Tibanna (feature coming soon!). A hybrid approach ensures the pipeline is accessible to all users.
Before getting started, we highly recommend reading through the usage section of each available sub command.
For more information about issues or trouble-shooting a problem, please checkout our FAQ prior to opening an issue on Github.
Snakemake and singularity must be installed on the target system. Snakemake orchestrates the execution of each step in the pipeline. To guarantee the highest level of reproducibility, each step relies on versioned images from DockerHub. Snakemake uses singaularity to pull these images onto the local filesystem prior to job execution, and as so, snakemake and singularity are the only two dependencies.
# XAVIER is configured to use different execution backends: local or slurm\n# view the help page for more information\nmodule load ccbrpipeliner\nxavier run --help\n\n# @slurm: uses slurm and singularity execution method\n# The slurm MODE will submit jobs to the cluster.\n# It is recommended running XAVIER in this mode.\n\n# Please note that you can dry-run the command below\n# by providing --runmode dryrun flag\n\n# Do not run this on the head node!\n# Grab an interactive node\nsinteractive --mem=110g --cpus-per-task=12 --gres=lscratch:200\nmodule load ccbrpipeliner\n\n# First, initialize the output directory\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode init\n\n# Second, do a dry run to visualize outputs\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode dryrun\n\n# Then do a complete run\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode run\n
"},{"location":"#frce","title":"FRCE","text":"
# grab an interactive node\nsrun --export all --pty --x11 bash\n\n# add xavier to path correctly\n. /mnt/projects/CCBR-Pipelines/pipelines/guis/latest/bin/setup\n\n# add SIF cache and TMPDIR path\nSIFCACHE=\"/mnt/projects/CCBR-Pipelines/SIFs/XAVIER\"\nTMPDIR=\"/scratch/cluster_scratch/$USER\"\n\n# run xavier\n\n# Initialize and then dryrun (or run)\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--sif-cache $SIFCACHE \\\n--tmp-dir $TMPDIR \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode init # run\n
This site is a living document, created for and by members like you. XAVIER is maintained by the members of CCBR and is improved by continuous feedback! We encourage you to contribute new content and make improvements to existing content via pull request to our repository.
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the \"Software\"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED \"AS IS\", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
Make sure you're keeping the changelog up-to-date during development. Ideally, every PR that includes a user-facing change (e.g. a new feature, bug fix, or any API change) should add a concise summary to the changelog with a link to the PR. Only approve or merge PRs that either update the changelog or have no user-facing changes.
"},{"location":"release-guide/#how-to-release-a-new-version-on-github","title":"How to release a new version on GitHub","text":"
Determine the new version number according to semantic versioning guidelines.
Update CHANGELOG.md:
Edit the heading for the development version to match the new version.
If needed, clean up the changelog -- fix any typos, optionally create subheadings for 'New features' and 'Bug fixes' if there are lots of changes, etc.
Update the version in src/__init__.py.
On GitHub, go to \"Releases\" and click \"Draft a new release\". https://github.com/CCBR/XAVIER/releases/new
Choose a tag: same as the version number.
Choose the target: most likely this should be the main branch, or a specific commit hash.
Set the title as the new version number, e.g. v3.0.2
Copy and paste the release notes from the CHANGELOG into the description box.
Check the box \"Set as the latest release\".
Click \"Publish release\".
Post release chores:
Add a new \"development version\" heading to the top of CHANGELOG.md.
Bump the version number in src/__init__.py to include -dev, e.g. v3.0.2-dev if you just released v3.0.2.
"},{"location":"release-guide/#how-to-install-a-release-on-biowulf","title":"How to install a release on biowulf","text":"
After releasing a new version on GitHub:
# go to the shared pipeline directory on biowulf\ncd /data/CCBR_Pipeliner/Pipelines/XAVIER\n\n# clone the new version tag (e.g. v3.0.2) to a hidden directory\ngit clone --depth 1 --branch v3.0.2 https://github.com/CCBR/XAVIER .v3.0.2\n\n# change permissions for the new directory so anyone will be able to use the pipeline\nchown -R :CCBR_Pipeliner .v3.0.2\nchmod -R a+rX /data/CCBR_Pipeliner/Pipelines/XAVIER/.v3.0.2\n\n# if needed, remove the old symlink for the minor version number\nrm -i v3.0\n\n# recreate the symlink to point to the new latest version\nln -s .v3.0.2 v3.0\n\n# you can verify that the symlink points to the new version with readlink\nreadlink -f v3.0\n
Versions of the ccbrpipeliner module only specify the major and minor version of each pipeline. If the new pipeline release only increments the patch number, ccbrpipeliner will use it automatically after you update the symlink as above. If you need to release a new major or minor version of a pipeline on biowulf, contact Kelly or Vishal.
Verify that ccbrpipeliner uses the latest version with:
Low-quality and adapters sequences are trimmed from the raw sequencing reads using Trimmomatic (v. 0.39)1. Trimmed reads are then aligned to the human hg38 reference genome using BWA mapping software (v. 0.7.17)2. Duplicate reads are marked using Samblaster (v. 0.1.25)3 and sorted using samtools (v. 1.8). Finally, base quality score recalibration is performed as indicated in the GATK4 (v. 4.2.2.0) best practices 4.
HaplotypeCaller from GATK4 (v. 4.2.2.0) is used to call germline variants, parallelized across chromosomes, and all samples in the cohort are joint genotyped together 4,5.
Somatic variant calling (SNPs and Indels) is performed using Mutect (v. 1.1.7)6, Mutect2 (GATK v. 4.2.0)7, Strelka2 (v. 2.9.0)8, and VarDict (v. 1.4)9 in tumor-normal mode. Variants from all callers are merged using the CombineVariants tool from GATK version 3.8-1. Genomic, functional and consequence annotations are added using Variant Effect Predictor (VEP v. 99)10 and converted to Mutation Annotation Format (MAF) using the vcf2maf tool (v. 1.6.16)11.
For Copy Number Variants (CNVs), Control-Freec (v. 11.6)12 is used to generate pileups, which are used as input for the R package 'sequenza' (v. 3.0.0)13. The complete Control-Freec workflow is then re-run using ploidy and cellularity estimates from 'sequenza'.
SOBDetector is a tool that scores variants based on strand-orientation bias, which can be a sign of DNA damage caused by fixation of tissue. This pipeline runs SOBDetector in a two-pass method. The first pass uses parameters provided with the software (calculated from publicly available data from TCGA), then cohort-specific bias metrics are computed from those results, and SOBDetector is re-run using these cohort-specific values.
"},{"location":"pipeline-details/methods/#quality-and-identity-metrics","title":"Quality and identity metrics","text":"
Ancestry and relatedness scores are generated using Somalier (v. 0.2.13)14. Contamination analyses are performed against viral and bacterial genomes from NCBI using Kraken2 (v. 2.1.2)15, as well as against mouse, human, and UniVec databases using FastQ Screen (v. 0.14.1)16. Sequence, mapping and variant statistics are computed using FastQC (v. 0.11.9), Qualimap (v. 2.2.1)17 and SNPeff (v. 4.3t)18. All of these metrics are combined into an interactive HTML report using MultiQC (v. 1.11)19.
Bolger, A.M., M. Lohse, and B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114-20.\u00a0\u21a9
Li, H. and R. Durbin, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009. 25(14): p. 1754-60.\u00a0\u21a9
Faust, G.G. and I.M. Hall, SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics, 2014. 30(17): p. 2503-5.\u00a0\u21a9
Van der Auwera, G.A. and B.D. O'Connor, Genomics in the cloud : using Docker, GATK, and WDL in Terra. First edition. ed. 2020, Sebastopol, CA: O'Reilly Media.\u00a0\u21a9\u21a9
Poplin, R., et al., Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv, 2018: p. 201178.\u00a0\u21a9
Cibulskis, K., et al., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol, 2013. 31(3): p. 213-9.\u00a0\u21a9
Benjamin, D., et al., Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 2019: p. 861054.\u00a0\u21a9
Kim, S., et al., Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods, 2018. 15(8): p. 591-594.\u00a0\u21a9
Lai, Z., et al., VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res, 2016. 44(11): p. e108.\u00a0\u21a9
McLaren, W., et al., The Ensembl Variant Effect Predictor. Genome Biol, 2016. 17(1): p. 122.\u00a0\u21a9
Memorial Sloan Kettering Cancer Center. vcf2maf. 2013; Available from: https://github.com/mskcc/vcf2maf.\u00a0\u21a9
Boeva, V., et al., Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics, 2012. 28(3): p. 423-5.\u00a0\u21a9
Favero, F., et al., Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol, 2015. 26(1): p. 64-70.\u00a0\u21a9
Pedersen, B. somalier: extract informative sites, evaluate relatedness, and perform quality-control on BAM/CRAM/BCF/VCF/GVCF. 2018; Available from: https://github.com/brentp/somalier.\u00a0\u21a9
Wood, D.E., J. Lu, and B. Langmead, Improved metagenomic analysis with Kraken 2. Genome Biol, 2019. 20(1): p. 257.\u00a0\u21a9
Wingett, S.W. and S. Andrews, FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res, 2018. 7: p. 1338.\u00a0\u21a9
Okonechnikov, K., A. Conesa, and F. Garcia-Alcalde, Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics, 2016. 32(2): p. 292-4.\u00a0\u21a9
Cingolani, P., et al., A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 2012. 6(2): p. 80-92.\u00a0\u21a9
Ewels, P., et al., MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 2016. 32(19): p. 3047-8.\u00a0\u21a9
Koster, J. and S. Rahmann, Snakemake-a scalable bioinformatics workflow engine. Bioinformatics, 2018. 34(20): p. 3600.\u00a0\u21a9
The output files and their locations are broken down here for the XAVIER pipeline. Pre-processing and germline variant calling steps are common but somatic variant calling is dependent on whether the pipeline was run in either (A) tumor-normal pair or (B) tumor-only analysis mode. All file locations are relative to the output directory specified during the job submission.
The output directory after a complete XAVIER run should look like:
xavier_output/\n\u251c\u2500\u2500 bams\n\u251c\u2500\u2500 cluster.json # cluster info for the run\n\u251c\u2500\u2500 config\n\u251c\u2500\u2500 config.json # config file for the run\n\u251c\u2500\u2500 fastqs\n\u251c\u2500\u2500 germline\n\u251c\u2500\u2500 indels.vcf.gz[.tbi] # raw germline INDELs\n\u251c\u2500\u2500 input_files\n\u251c\u2500\u2500 intervals.list\n\u251c\u2500\u2500 {sample1}-normal.R1.fastq.gz -> /path/to/{sample1}-normal.R1.fastq.gz\n\u251c\u2500\u2500 {sample1}-normal.R2.fastq.gz -> /path/to/{sample1}-normal.R2.fastq.gz\n\u251c\u2500\u2500 {sample1}-tumor.R1.fastq.gz -> /path/to/{sample1}-tumor.R1.fastq.gz\n\u251c\u2500\u2500 {sample1}-tumor.R2.fastq.gz -> /path/to/{sample1}-tumor.R2.fastq.gz\n.\n.\n.\n\u251c\u2500\u2500 kickoff.sh\n\u251c\u2500\u2500 logfiles\n\u251c\u2500\u2500 QC\n\u251c\u2500\u2500 resources\n\u251c\u2500\u2500 snps.vcf.gz[.tbi] # raw germline SNPs\n\u251c\u2500\u2500 somatic_paired # in case of tumor-normal paired run\n\u251c\u2500\u2500 somatic_tumor_only # in case of tumor-only run\n\u2514\u2500\u2500 workflow\n
Below we describe the different folders that contain specific outputs obtained for all samples from the XAVIER pipeline
The QC folder contains all the Quality-Control analyses performed at different steps of the pipeline for each sample to assess sequencing quality before and after adapter trimming, microbial taxonomic composition, contamination, variant calling, etc. The final summary report and data is available finalQC folder. \\ The MultiQC report also contains results from other analysis like mapping statistics, ancestry and relatedness, etc. It is recommended to study the MultiQC report first to get a birds eye view of the sequence data quality.
The bams folder contain two subfolders chrom_split and final_bams. final_bams contains the final processed BAM files for each sample in the run and the chrom_split folder contains all the sample BAM files split by each chromosome.
This folder contains the output from the GATK Best Practices pipeline to obtain germline variants with a few alterations detailed below. Briefly, joint SNP and INDEL variant detection is conducted across all samples included in a pipeline run using the GATK Haplotypcaller under default settings. Raw variants are then subsequently filtered based on several GATK annotations: \\ A strict set of criteria (QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < -12.5, ReadPosRankSum < -8.0 for SNPs; QD < 2.0, FS > 200.0, ReadPosRankSum < -20.0 for INDELs) generates the 'combined.strictFilter.vcf'.
This call set is highly stringent, maximizing the true positive rate at the expense of an elevated false negative rate. This call set is really only intended for more general population genetic scale analyses (e.g., burden tests, admixture, linkage/pedigree based analysis, etc.) where false positives can be significantly confounding.
In case of human sequence data, a basic analyses of sample relatedness and ancestry (e.g., % European, African, etc.) is also performed using somalier.
The output folder looks like:
germline/\n\u251c\u2500\u2500 gVCFs\n.\n.\n.\n\u251c\u2500\u2500 somalier # only for hg38 genome\n\u2514\u2500\u2500 VCF\n
The VCF folder contains the final filtered germline variants (SNPs and INDELs) for all samples combined. The folder also contains raw variants for each sample, all samples combined, and also combined raw variants split by chromosome.
This folder contains the snakemake log files and computational statistics for the XAVIER run. All the log files (i.e., standard output and error) for each individual step are in the slurmfiles folder. These logfiles are important to diagnose errors in case the pipeline fails.
This workflow calls somatic SNPs and INDELs using multiple variant detection algorithms. For each of these tools, variants are called in a paired tumor-normal fashion, with default settings. See Pipeline Details for more information about the tools used and their parameter settings.
For each sample, the resulting VCF is fully annotated using VEP and converted to a MAF file using the vcf2maf tool. Resulting MAF files are found in maf folder within each caller's results directory (i.e., mutect2_out, strelka_out, etc.). Individual sample MAF files are then merged and saved in merged_somatic_variants directory.
For Mutect2, we use a panel of normals (PON) developed from the ExAC (excluding TCGA) dataset, filtered for variants <0.001 in the general population, and also including and in-house set of blacklisted recurrent germline variants that are not found in any population databases.
For Copy Number Variants (CNVs), two tools are employed in tandem. First, Control-FREEC is run with default parameters. This generates pileup files that can be used by Sequenza, primarily for jointly estimating contamination and ploidy. These value are used to run Freec a second time for improved performance.
In general, the tumor-only pipeline is a stripped down version of the tumor-normal pipeline. We only run MuTect2, Mutect, and VarDict for somatic variant detection, with the same PON and filtering as described above for the tumor-normal pipeline.
Workflow diagram of the XAVIER: the pipeline is composed of a series of data processing steps to trim, align, and recalibrate reads prior to calling variants. These data processing steps closely follow GATK's best pratices for cleaning up raw alignments. The pipeline also consists of a series of comprehensive quality-control steps.
Removes variants with certain flags from vardict; (1) Germline: detected in germline sample (pass all quality parameters); (2) LikelyLOH: detected in germline but either lost in tumor OR 20-80% in germline, but increased to 1-opt_V (95%); (3) AFDiff: detected in tumor (pass quality parameters) and present in germline but didn\u2019t pass quality parameters.
varscan
pileup
-d 100000 -q 15 -Q 15
samtools mpileup arguments; max depth of 100,000; min mapping quality of 15; min base quality of 15
calling
--strand-filter 0
Do not ignore variants with >90% support on one strand
--min-var-freq 0.01
Minimum variant allele frequency threshold 0.01
--output-vcf 1
Output in VCF format
--variants 1
Report only variant (SNP/indel) positions
all
GATK SelectVariants
--exclude-filtered
Removes non-PASS variants
--discordance
Remove variants found in supplied file (same as panel-of-normals file)
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier cache sub command in more detail. With minimal configuration, the cache sub command enables you to cache remote resources for the xavier pipeline. Caching remote resources allows the pipeline to run in an offline mode. The cache sub command can also be used to pull our pre-built reference bundles onto a new cluster or target system.
The cache sub command creates local cache on the filesysytem for resources hosted on DockerHub or AWS S3. These resources are normally pulled onto the filesystem when the pipeline runs; however, due to network issues or DockerHub pull rate limits, it may make sense to pull the resources once so a shared cache can be created and re-used. It is worth noting that a singularity cache cannot normally be shared across users. Singularity strictly enforces that its cache is owned by the user. To get around this issue, the cache subcommand can be used to create local SIFs on the filesystem from images on DockerHub.
XAVIER pipeline can be executed from either using the graphical user interface (GUI) or the command line interface (CLI). GUI offers a more interactive way for the user to provide input and adjust parameter settings. This part of the documentation describes how to run xavier using the GUI (with screenshots). See Command Line tab to read more about the xavier executable and running XAVIER pipeline using the CLI.
"},{"location":"usage/gui/#2-setting-up-xavier","title":"2. Setting up XAVIER","text":""},{"location":"usage/gui/#21-login-to-cluster","title":"2.1 Login to cluster","text":"
# Setup Step 1.) ssh into cluster's head node\n# example below for Biowulf cluster\nssh -Y $USER@biowulf.nih.gov\n
"},{"location":"usage/gui/#22-grab-an-interactive-node","title":"2.2 Grab an interactive node","text":"
# Setup Step 2.) Please do not run XAVIER on the head node!\n# Grab an interactive node first\nsinteractive --time=12:00:00 --mem=8gb --cpus-per-task=4\n
NOTE: ccbrpipeliner is a custom module created on biowulf which contains various NGS data analysis pipelines developed, tested, and benchmarked by experts at CCBR.
# Setup Step 3.) Add ccbrpipeliner module\nmodule purge # to reset the module environment\nmodule load ccbrpipeliner\n
If the module was loaded correctly, the greetings message should be displayed.
[+] Loading ccbrpipeliner 5 ...\n###########################################################################\n CCBR Pipeliner\n###########################################################################\n \"ccbrpipeliner\" is a suite of end-to-end pipelines and tools\n Visit https://github.com/ccbr for more details.\n Pipelines are available on BIOWULF and FRCE.\n Tools are available on BIOWULF, HELIX and FRCE.\n\n The following pipelines/tools will be loaded in this module:\n\n RENEE v2.5 https://ccbr.github.io/RENEE/\n XAVIER v3.0 https://ccbr.github.io/XAVIER/\n CARLISLE v2.4 https://ccbr.github.io/CARLISLE/\n CHAMPAGNE v0.2 https://ccbr.github.io/CHAMPAGNE/\n CRUISE v0.1 https://ccbr.github.io/CRUISE/\n\n spacesavers2 v0.10 https://ccbr.github.io/spacesavers2/\n permfix v0.6 https://github.com/ccbr/permfix\n###########################################################################\nThank you for using CCBR Pipeliner\n###########################################################################\n
To run the XAVIER pipeline from the GUI, simply enter:
xavier gui\n
and it will launch the XAVIER window.
Note: Please wait until window created! message appears on the terminal.
"},{"location":"usage/gui/#32-folder-paths-and-reference-genomes","title":"3.2 Folder paths and reference genomes","text":"
To enter the location of the input folder containing FASTQ files and the location where the output folders should be created, either simply type the absolute paths
or use the Browse tab to choose the input and output directories
Next, from the drop down menu select the reference genome (hg38/mm10)
In case the paired normal samples are unavailable, XAVIER pipeline can be run in tumor-only mode which does not require paired samples information. However, in the absence of matching normal samples, CNV analysis is also unavailable.
After all the information is filled out, press Submit.
If the pipeline detects no errors and the run was submitted, a new window appears that has the output of a \"dry-run\" which summarizes each step of the pipeline.
Click OK
A dialogue box will popup to confirm submitting the job to slurm.
Click Yes
The dry-run output will be displayed again and the master job will be submitted. An email notification will be sent out when the pipeline starts and ends.
The XAVIER gui will ask to submit another job.
Click Yes to start again or No to close the XAVIER gui.
Users can input certain additional settings for the pipeline run including running an additional step to correct strand orientation bias in Formalin-Fixed Paraffin-Embedded (FFPE) samples and to provide a custom exome targets BED file. This file can be obtained from the manufacturer of the target capture kit that was used.
"},{"location":"usage/gui/#4-special-instructions-for-biowulf","title":"4. Special instructions for Biowulf","text":"
XAVIER GUI natively uses the X11 Window System to run XAVIER pipeline and display the graphics on a personal desktop or laptop. However, if running XAVIER specifically on NIH's Biowulf cluster, the HPC staff recommends NoMachine (NX) to run graphics applications.
Please see details here on how to install and connect to Biowulf on your local computer using NoMachine.
Once connected to Biowulf using NX, right click to open a terminal connection
and start an interactive session.
Similar to the instructions above, load ccbrpipeliner module and enter xavier gui to launch the XAVIER gui.
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier run sub command in more detail. With minimal configuration, the run sub command enables you to start running xavier pipeline.
Setting up the xavier pipeline is fast and easy! In its most basic form, xavier run only has four required inputs.
The synopsis for each command shows its parameters and their usage. Optional parameters are shown in square brackets.
A user must provide a list of FastQ or BAM files (globbing is supported) to analyze via --input argument, an output directory to store results via --output argument, an exome targets BED file for the samples' capture kit, and select reference genome for alignment and annotation via the --genome argument.
Use you can always use the -h option for information on a specific command.
Each of the following arguments are required. Failure to provide a required argument will result in a non-zero exit-code.
--input INPUT [INPUT ...]
Input FastQ or BAM file(s) to process. type: file(s)
One or more FastQ files can be provided. The pipeline does NOT support single-end WES data. Please provide either a set of FastQ files or a set of BAM files. The pipeline does NOT support processing a mixture of FastQ files and BAM files. From the command-line, each input file should separated by a space. Globbing is supported! This makes selecting FastQ files easy. Input FastQ files should be gzipp-ed.
This location is where the pipeline will create all of its output files, also known as the pipeline's working directory. If the provided output directory does not exist, it will be initialized automatically.
Example: --output /data/$USER/WES_hg38
--runmode {init,dryrun,run} `
Execution Process. type: string
User should initialize the pipeline folder by first running --runmode init User should then perform a dry-run to list all steps the pipeline will take--runmode dryrun User should then perform the full run --runmode run
Example: --runmode init THEN --runmode dryrun THEN --runmode run
--genome {hg38, custom.json}
Reference genome. type: string/file
This option defines the reference genome for your set of samples. On Biowulf, xavier does comes bundled with pre built reference files for human samples; however, it is worth noting that the pipeline does accept a pre-built resource bundle pulled with the cache sub command (coming soon). Currently, the pipeline only supports the human reference hg38; however, support for mouse reference mm10 will be added soon.
Pre built Option Here is a list of available pre built genomes on Biowulf: hg38, mm10.
Custom Option For users running the pipeline outside of Biowulf, a pre-built resource bundle can be pulled with the cache sub command (coming soon). Please supply the custom reference JSON file that was generated by the cache sub command.
Example: --genome hg38 OR --genome /data/${USER}/hg38/hg38.json
--targets TARGETS
Exome targets BED file. type: file
This file can be obtained from the manufacturer of the target capture kit that was used.
If not provided, the default targets file from the genome config is used
Each of the following arguments are optional and do not need to be provided.
-h, --help
Display Help. type: boolean flag
Shows command's synopsis, help message, and an example command
Example: --help
--silent
Silence standard output. type: boolean flag
Reduces the amount of information directed to standard output when submitting master job to the job scheduler. Only the job id of the master job is returned.
Example: --silent
--mode {local,slurm}
Execution Method. type: string default: slurm
Execution Method. Defines the mode or method of execution. Valid mode options include: local or slurm.
local Local executions will run serially on compute instance. This is useful for testing, debugging, or when a users does not have access to a high performance computing environment. If this option is not provided, it will default to a local execution mode.
slurm The slurm execution method will submit jobs to a cluster using a singularity backend. It is recommended running xavier in this mode as execution will be significantly faster in a distributed environment.
Example: --mode slurm
--job-name JOB_NAME
Set the name of the pipeline's master job. type: string > default: pl:xavier
When submitting the pipeline to a job scheduler, like SLURM, this option always you to set the name of the pipeline's master job. By default, the name of the pipeline's master job is set to \"pl:xavier\".
List of variant callers to detect mutations. Please select from one or more of the following options: [mutect2, mutect, strelka, vardict, varscan]. Defaults to using all variant callers.
Example: --callers mutect2 strelka varscan
--pairs PAIRS
Tumor normal pairs file. type: file
This tab delimited file contains two columns with the names of tumor and normal pairs, one per line. The header of the file needs to be Tumor for the tumor column and Normal for the normal column. The base name of each sample should be listed in the pairs file. The base name of a given sample can be determined by removing the following extension from the sample's R1 FastQ file: .R1.fastq.gz. Contents of example pairs file:
Normal Tumor\nSample4_CRL1622_S31 Sample10_ARK1_S37\nSample4_CRL1622_S31 Sample11_ACI_158_S38\n
Example: --pairs /data/$USER/pairs.tsv
--ffpe
Apply FFPE correction. type: boolean flag
Runs an additional steps to correct strand orientation bias in Formalin-Fixed Paraffin-Embedded (FFPE) samples. Do NOT use this option with non-FFPE samples.
Example: --ffpe
--cnv
Call copy number variations (CNVs). type: boolean flag
CNVs will only be called from tumor-normal pairs. If this option is provided without providing a --pairs file, CNVs will NOT be called.
Example: --cnv
--singularity-cache SINGULARITY_CACHE
Overrides the $SINGULARITY_CACHEDIR environment variable. type: path default: --output OUTPUT/.singularity
Singularity will cache image layers pulled from remote registries. This ultimately speeds up the process of pull an image from DockerHub if an image layer already exists in the singularity cache directory. By default, the cache is set to the value provided to the --output argument. Please note that this cache cannot be shared across users. Singularity strictly enforces you own the cache directory and will return a non-zero exit code if you do not own the cache directory! See the --sif-cache option to create a shareable resource.
Path where a local cache of SIFs are stored. type: path
Uses a local cache of SIFs on the filesystem. This SIF cache can be shared across users if permissions are set correctly. If a SIF does not exist in the SIF cache, the image will be pulled from Dockerhub and a warning message will be displayed. The xavier cache subcommand can be used to create a local SIF cache. Please see xavier cache for more information. This command is extremely useful for avoiding DockerHub pull rate limits. It also remove any potential errors that could occur due to network issues or DockerHub being temporarily unavailable. We recommend running xavier with this option when ever possible.
Example: --singularity-cache /data/$USER/SIFs
--threads THREADS
Max number of threads for each process. type: int default: 2
Max number of threads for each process. This option is more applicable when running the pipeline with --mode local. It is recommended setting this value to the maximum number of CPUs available on the host machine.
# Step 1.) Grab an interactive node\n# Do not run on head node!\nsinteractive --mem=8g --cpus-per-task=4\nmodule purge\nmodule load ccbrpipeliner\n\n# Step 2A.) Initialize the all resources to the output folder\nxavier run --input tests/data/*.R?.fastq.gz \\\n --output /data/$USER/xavier_hg38 \\\n --genome hg38 \\\n --targets Agilent_SSv7_allExons_hg38.bed \\\n --mode slurm \\\n --runmode init\n\n# Step 2B.) Dry-run the pipeline\nxavier run --input tests/data/*.R?.fastq.gz \\\n --output /data/$USER/xavier_hg38 \\\n --genome hg38 \\\n --targets Agilent_SSv7_allExons_hg38.bed \\\n --mode slurm \\\n --runmode dryrun\n\n# Step 2C.) Run the XAVIER pipeline\n# The slurm mode will submit jobs to the cluster.\n# It is recommended running xavier in this mode.\nxavier run --input tests/data/*.R?.fastq.gz \\\n --output /data/$USER/xavier_hg38 \\\n --genome hg38 \\\n --targets Agilent_SSv7_allExons_hg38.bed \\\n --mode slurm \\\n --runmode run\n
The example dataset in tests/data in this repository is a very small subsampled dataset, and some steps of the pipeline fail due to the small size (CNV callling, somalier, etc). We have a larger subsample (25% of a full human dataset) available on Biowulf if you would like to test the full functionality of the pipeline: /data/CCBR_Pipeliner/testdata/XAVIER/human_subset/*.R?.fastq.gz
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier unlock sub command in more detail. With minimal configuration, the unlock sub command enables you to unlock a pipeline output directory.
If the pipeline fails ungracefully, it maybe required to unlock the working directory before proceeding again. Snakemake will inform a user when it maybe necessary to unlock a working directory with an error message stating: Error: Directory cannot be locked.
Please verify that the pipeline is not running before running this command. If the pipeline is currently running, the workflow manager will report the working directory is locked. The is the default behavior of snakemake, and it is normal. Do NOT run this command if the pipeline is still running! Please kill the master job and it's child jobs prior to running this command.
Unlocking xavier pipeline output directory is fast and easy! In its most basic form, xavier unlock only has one required input.
The synopsis for this command shows its parameters and their usage. Optional parameters are shown in square brackets.
A user must provide an output directory to unlock via --output argument. After running the unlock sub command, you can resume the build or run pipeline from where it left off by re-running it.
Use you can always use the -h option for information on a specific command.
Path to a previous run's output directory. This will remove a lock on the working directory. Please verify that the pipeline is not running before running this command. Example: --output /data/$USER/WES_hg38
# Step 0.) Grab an interactive node (do not run on head node)\nsinteractive --mem=8g -N 1 -n 4\nmodule purge\nmodule load ccbrpipeliner\n\n# Step 1.) Unlock a pipeline output directory\nxavier unlock --output /data/$USER/xavier_hg38\n
"}]}
\ No newline at end of file
+{"config":{"lang":["en"],"separator":"[\\s\\-]+","pipeline":["stopWordFilter"]},"docs":[{"location":"","title":"About","text":""},{"location":"#xavier-exome-analysis-and-variant-explorer","title":"XAVIER - eXome Analysis and Variant explorER \ud83d\udd2c","text":"
_XAVIER - eXome Analysis and Variant explorER_. This is the home of the pipeline, XAVIER. Its long-term goals: to accurately call germline and somatic variants, to infer CNVs, and to boldly annotate variants like no pipeline before!
Welcome to XAVIER! Before getting started, we highly recommend reading through xavier's documentation.
The xavier pipeline is composed several inter-related sub commands to setup and run the pipeline across different systems. Each of the available sub commands perform different functions:
xavier run: Run the XAVIER pipeline with your input files.
xavier unlock: Unlocks a previous runs output directory.
XAVIER is a comprehensive whole exome-sequencing pipeline following the Broad's set of best practices. It relies on technologies like Singularity1 to maintain the highest-level of reproducibility. The pipeline consists of a series of data processing and quality-control steps orchestrated by Snakemake2, a flexible and scalable workflow management system, to submit jobs to a cluster or cloud provider.
The pipeline is compatible with data generated from Illumina short-read sequencing technologies. As input, it accepts a set of FastQ or BAM files and can be run locally on a compute instance, on-premise using a cluster, or on the cloud (feature coming soon!). A user can define the method or mode of execution. The pipeline can submit jobs to a cluster using a job scheduler like SLURM, or run on AWS using Tibanna (feature coming soon!). A hybrid approach ensures the pipeline is accessible to all users.
Before getting started, we highly recommend reading through the usage section of each available sub command.
For more information about issues or trouble-shooting a problem, please checkout our FAQ prior to opening an issue on Github.
Snakemake and singularity must be installed on the target system. Snakemake orchestrates the execution of each step in the pipeline. To guarantee the highest level of reproducibility, each step relies on versioned images from DockerHub. Snakemake uses singaularity to pull these images onto the local filesystem prior to job execution, and as so, snakemake and singularity are the only two dependencies.
# XAVIER is configured to use different execution backends: local or slurm\n# view the help page for more information\nmodule load ccbrpipeliner\nxavier run --help\n\n# @slurm: uses slurm and singularity execution method\n# The slurm MODE will submit jobs to the cluster.\n# It is recommended running XAVIER in this mode.\n\n# Please note that you can dry-run the command below\n# by providing --runmode dryrun flag\n\n# Do not run this on the head node!\n# Grab an interactive node\nsinteractive --mem=110g --cpus-per-task=12 --gres=lscratch:200\nmodule load ccbrpipeliner\n\n# First, initialize the output directory\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode init\n\n# Second, do a dry run to visualize outputs\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode dryrun\n\n# Then do a complete run\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode run\n
"},{"location":"#frce","title":"FRCE","text":"
# grab an interactive node\nsrun --export all --pty --x11 bash\n\n# add xavier to path correctly\n. /mnt/projects/CCBR-Pipelines/pipelines/guis/latest/bin/setup\n\n# add SIF cache and TMPDIR path\nSIFCACHE=\"/mnt/projects/CCBR-Pipelines/SIFs/XAVIER\"\nTMPDIR=\"/scratch/cluster_scratch/$USER\"\n\n# run xavier\n\n# Initialize and then dryrun (or run)\nxavier run \\\n--input data/*.R?.fastq.gz \\\n--output /data/$USER/xavier_hg38 \\\n--genome hg38 \\\n--sif-cache $SIFCACHE \\\n--tmp-dir $TMPDIR \\\n--pairs pairs.txt \\\n--targets resources/Agilent_SSv7_allExons_hg38.bed \\\n--mode slurm \\\n--runmode init # run\n
This site is a living document, created for and by members like you. XAVIER is maintained by the members of CCBR and is improved by continuous feedback! We encourage you to contribute new content and make improvements to existing content via pull request to our repository.
1. Kurtzer GM, Sochat V, Bauer MW (2017). Singularity: Scientific containers for mobility of compute. PLoS ONE 12(5): e0177459.2. Koster, J. and S. Rahmann (2018). \"Snakemake-a scalable bioinformatics workflow engine.\" Bioinformatics 34(20): 3600.
"},{"location":"contributing/","title":"Contributing to XAVIER","text":""},{"location":"contributing/#proposing-changes-with-issues","title":"Proposing changes with issues","text":"
If you want to make a change, it's a good idea to first open an issue and make sure someone from the team agrees that it\u2019s needed.
If you've decided to work on an issue, assign yourself to the issue so others will know you're working on it.
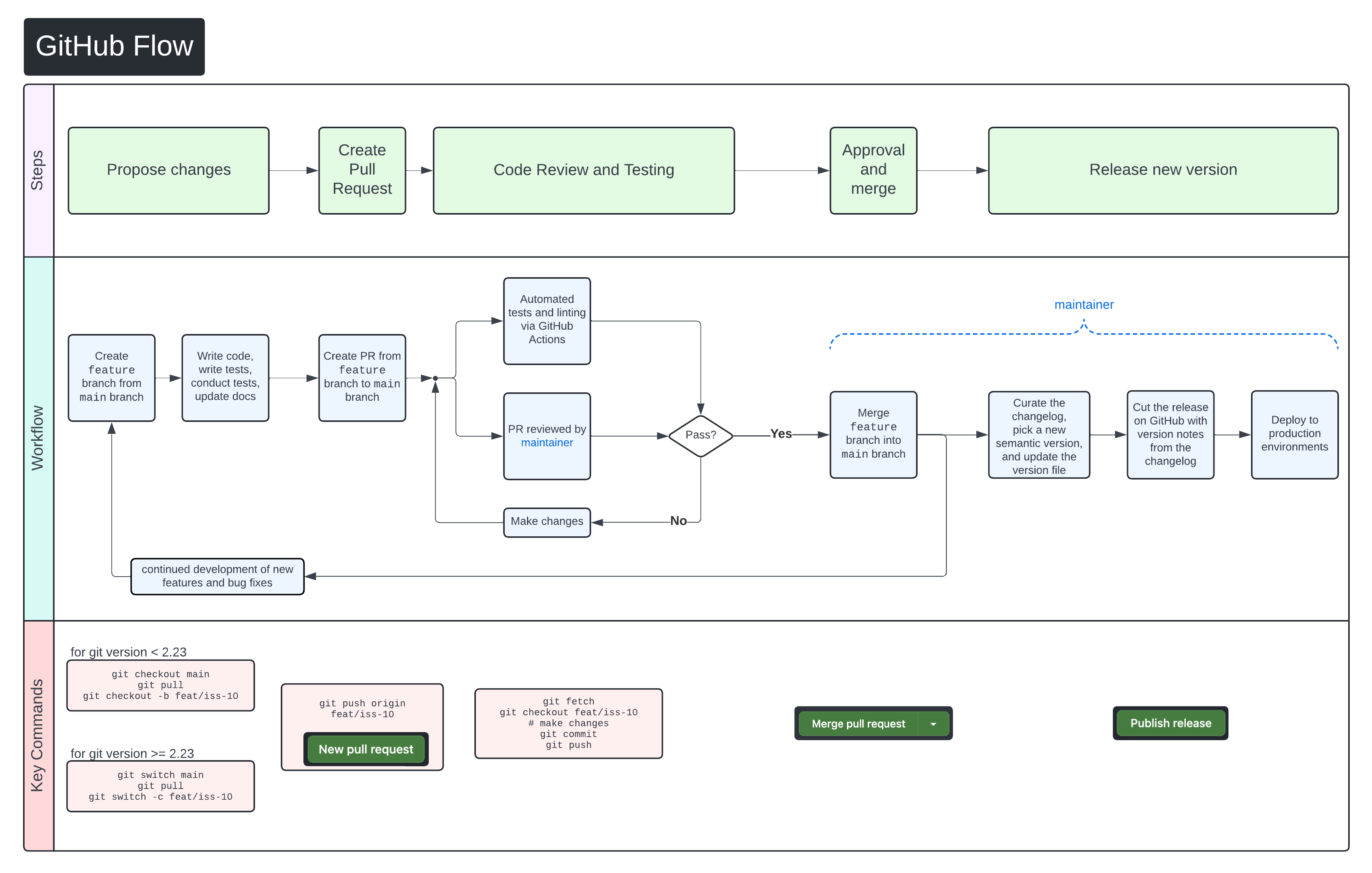
We use GitHub Flow as our collaboration process. Follow the steps below for detailed instructions on contributing changes to XAVIER.
"},{"location":"contributing/#clone-the-repo","title":"Clone the repo","text":"
If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
"},{"location":"contributing/#if-this-is-your-first-time-cloning-the-repo-you-may-need-to-install-dependencies","title":"If this is your first time cloning the repo, you may need to install dependencies","text":"
Install snakemake and singularity or docker if needed (biowulf already has these available as modules).
Install the python dependencies with pip
pip install .\n
If you're developing on biowulf, you can use our shared conda environment which already has these dependencies installed
Install pre-commit if you don't already have it. Then from the repo's root directory, run
pre-commit install\n
This will install the repo's pre-commit hooks. You'll only need to do this step the first time you clone the repo.
"},{"location":"contributing/#create-a-branch","title":"Create a branch","text":"
Create a Git branch for your pull request (PR). Give the branch a descriptive name for the changes you will make, such as iss-10 if it is for a specific issue.
# create a new branch and switch to it\ngit branch iss-10\ngit switch iss-10\n
Switched to a new branch 'iss-10'
"},{"location":"contributing/#make-your-changes","title":"Make your changes","text":"
Edit the code, write and run tests, and update the documentation as needed.
Changes to the python package code will also need unit tests to demonstrate that the changes work as intended. We write unit tests with pytest and store them in the tests/ subdirectory. Run the tests with python -m pytest.
If you change the workflow, please run the workflow with the test profile and make sure your new feature or bug fix works as intended.
If you have added a new feature or changed the API of an existing feature, you will likely need to update the documentation in docs/.
"},{"location":"contributing/#commit-and-push-your-changes","title":"Commit and push your changes","text":"
If you're not sure how often you should commit or what your commits should consist of, we recommend following the \"atomic commits\" principle where each commit contains one new feature, fix, or task. Learn more about atomic commits here: https://www.freshconsulting.com/insights/blog/atomic-commits/
First, add the files that you changed to the staging area:
git add path/to/changed/files/\n
Then make the commit. Your commit message should follow the Conventional Commits specification. Briefly, each commit should start with one of the approved types such as feat, fix, docs, etc. followed by a description of the commit. Take a look at the Conventional Commits specification for more detailed information about how to write commit messages.
git commit -m 'feat: create function for awesome feature'\n
pre-commit will enforce that your commit message and the code changes are styled correctly and will attempt to make corrections if needed.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Failed
hook id: trailing-whitespace
exit code: 1
files were modified by this hook > Fixing path/to/changed/files/file.txt > codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped
In the example above, one of the hooks modified a file in the proposed commit, so the pre-commit check failed. You can run git diff to see the changes that pre-commit made and git status to see which files were modified. To proceed with the commit, re-add the modified file(s) and re-run the commit command:
git add path/to/changed/files/file.txt\ngit commit -m 'feat: create function for awesome feature'\n
This time, all the hooks either passed or were skipped (e.g. hooks that only run on R code will not run if no R files were committed). When the pre-commit check is successful, the usual commit success message will appear after the pre-commit messages showing that the commit was created.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Passed codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped Conventional Commit......................................................Passed > [iss-10 9ff256e] feat: create function for awesome feature 1 file changed, 22 insertions(+), 3 deletions(-)
Finally, push your changes to GitHub:
git push\n
If this is the first time you are pushing this branch, you may have to explicitly set the upstream branch:
git push --set-upstream origin iss-10\n
Enumerating objects: 7, done. Counting objects: 100% (7/7), done. Delta compression using up to 10 threads Compressing objects: 100% (4/4), done. Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done. Total 4 (delta 3), reused 0 (delta 0), pack-reused 0 remote: Resolving deltas: 100% (3/3), completed with 3 local objects. remote: remote: Create a pull request for 'iss-10' on GitHub by visiting: remote: https://github.com/CCBR/XAVIER/pull/new/iss-10 remote: To https://github.com/CCBR/XAVIER > > [new branch] iss-10 -> iss-10 branch 'iss-10' set up to track 'origin/iss-10'.
We recommend pushing your commits often so they will be backed up on GitHub. You can view the files in your branch on GitHub at https://github.com/CCBR/XAVIER/tree/<your-branch-name> (replace <your-branch-name> with the actual name of your branch).
"},{"location":"contributing/#create-the-pr","title":"Create the PR","text":"
Once your branch is ready, create a PR on GitHub: https://github.com/CCBR/XAVIER/pull/new/
Select the branch you just pushed:
Edit the PR title and description. The title should briefly describe the change. Follow the comments in the template to fill out the body of the PR, and you can delete the comments (everything between <!-- and -->) as you go. Be sure to fill out the checklist, checking off items as you complete them or striking through any irrelevant items. When you're ready, click 'Create pull request' to open it.
Optionally, you can mark the PR as a draft if you're not yet ready for it to be reviewed, then change it later when you're ready.
"},{"location":"contributing/#wait-for-a-maintainer-to-review-your-pr","title":"Wait for a maintainer to review your PR","text":"
We will do our best to follow the tidyverse code review principles: https://code-review.tidyverse.org/. The reviewer may suggest that you make changes before accepting your PR in order to improve the code quality or style. If that's the case, continue to make changes in your branch and push them to GitHub, and they will appear in the PR.
Once the PR is approved, the maintainer will merge it and the issue(s) the PR links will close automatically. Congratulations and thank you for your contribution!
"},{"location":"contributing/#after-your-pr-has-been-merged","title":"After your PR has been merged","text":"
After your PR has been merged, update your local clone of the repo by switching to the main branch and pulling the latest changes:
git checkout main\ngit pull\n
It's a good idea to run git pull before creating a new branch so it will start from the most recent commits in main.
"},{"location":"contributing/#helpful-links-for-more-information","title":"Helpful links for more information","text":"
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the \"Software\"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED \"AS IS\", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
Make sure you're keeping the changelog up-to-date during development. Ideally, every PR that includes a user-facing change (e.g. a new feature, bug fix, or any API change) should add a concise summary to the changelog with a link to the PR. Only approve or merge PRs that either update the changelog or have no user-facing changes.
"},{"location":"release-guide/#how-to-release-a-new-version-on-github","title":"How to release a new version on GitHub","text":"
Determine the new version number according to semantic versioning guidelines.
Update CHANGELOG.md:
Edit the heading for the development version to match the new version.
If needed, clean up the changelog -- fix any typos, optionally create subheadings for 'New features' and 'Bug fixes' if there are lots of changes, etc.
Update the version in src/__init__.py.
On GitHub, go to \"Releases\" and click \"Draft a new release\". https://github.com/CCBR/XAVIER/releases/new
Choose a tag: same as the version number.
Choose the target: most likely this should be the main branch, or a specific commit hash.
Set the title as the new version number, e.g. v3.0.2
Copy and paste the release notes from the CHANGELOG into the description box.
Check the box \"Set as the latest release\".
Click \"Publish release\".
Post release chores:
Add a new \"development version\" heading to the top of CHANGELOG.md.
Bump the version number in src/__init__.py to include -dev, e.g. v3.0.2-dev if you just released v3.0.2.
"},{"location":"release-guide/#how-to-install-a-release-on-biowulf","title":"How to install a release on biowulf","text":"
After releasing a new version on GitHub:
# go to the shared pipeline directory on biowulf\ncd /data/CCBR_Pipeliner/Pipelines/XAVIER\n\n# clone the new version tag (e.g. v3.0.2) to a hidden directory\ngit clone --depth 1 --branch v3.0.2 https://github.com/CCBR/XAVIER .v3.0.2\n\n# change permissions for the new directory so anyone will be able to use the pipeline\nchown -R :CCBR_Pipeliner .v3.0.2\nchmod -R a+rX /data/CCBR_Pipeliner/Pipelines/XAVIER/.v3.0.2\n\n# if needed, remove the old symlink for the minor version number\nrm -i v3.0\n\n# recreate the symlink to point to the new latest version\nln -s .v3.0.2 v3.0\n\n# you can verify that the symlink points to the new version with readlink\nreadlink -f v3.0\n
Versions of the ccbrpipeliner module only specify the major and minor version of each pipeline. If the new pipeline release only increments the patch number, ccbrpipeliner will use it automatically after you update the symlink as above. If you need to release a new major or minor version of a pipeline on biowulf, contact Kelly or Vishal.
Verify that ccbrpipeliner uses the latest version with:
Low-quality and adapters sequences are trimmed from the raw sequencing reads using Trimmomatic (v. 0.39)1. Trimmed reads are then aligned to the human hg38 reference genome using BWA mapping software (v. 0.7.17)2. Duplicate reads are marked using Samblaster (v. 0.1.25)3 and sorted using samtools (v. 1.8). Finally, base quality score recalibration is performed as indicated in the GATK4 (v. 4.2.2.0) best practices 4.
HaplotypeCaller from GATK4 (v. 4.2.2.0) is used to call germline variants, parallelized across chromosomes, and all samples in the cohort are joint genotyped together 4,5.
Somatic variant calling (SNPs and Indels) is performed using Mutect (v. 1.1.7)6, Mutect2 (GATK v. 4.2.0)7, Strelka2 (v. 2.9.0)8, and VarDict (v. 1.4)9 in tumor-normal mode. Variants from all callers are merged using the CombineVariants tool from GATK version 3.8-1. Genomic, functional and consequence annotations are added using Variant Effect Predictor (VEP v. 99)10 and converted to Mutation Annotation Format (MAF) using the vcf2maf tool (v. 1.6.16)11.
For Copy Number Variants (CNVs), Control-Freec (v. 11.6)12 is used to generate pileups, which are used as input for the R package 'sequenza' (v. 3.0.0)13. The complete Control-Freec workflow is then re-run using ploidy and cellularity estimates from 'sequenza'.
SOBDetector is a tool that scores variants based on strand-orientation bias, which can be a sign of DNA damage caused by fixation of tissue. This pipeline runs SOBDetector in a two-pass method. The first pass uses parameters provided with the software (calculated from publicly available data from TCGA), then cohort-specific bias metrics are computed from those results, and SOBDetector is re-run using these cohort-specific values.
"},{"location":"pipeline-details/methods/#quality-and-identity-metrics","title":"Quality and identity metrics","text":"
Ancestry and relatedness scores are generated using Somalier (v. 0.2.13)14. Contamination analyses are performed against viral and bacterial genomes from NCBI using Kraken2 (v. 2.1.2)15, as well as against mouse, human, and UniVec databases using FastQ Screen (v. 0.14.1)16. Sequence, mapping and variant statistics are computed using FastQC (v. 0.11.9), Qualimap (v. 2.2.1)17 and SNPeff (v. 4.3t)18. All of these metrics are combined into an interactive HTML report using MultiQC (v. 1.11)19.
Bolger, A.M., M. Lohse, and B. Usadel, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 2014. 30(15): p. 2114-20.\u00a0\u21a9
Li, H. and R. Durbin, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009. 25(14): p. 1754-60.\u00a0\u21a9
Faust, G.G. and I.M. Hall, SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics, 2014. 30(17): p. 2503-5.\u00a0\u21a9
Van der Auwera, G.A. and B.D. O'Connor, Genomics in the cloud : using Docker, GATK, and WDL in Terra. First edition. ed. 2020, Sebastopol, CA: O'Reilly Media.\u00a0\u21a9\u21a9
Poplin, R., et al., Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv, 2018: p. 201178.\u00a0\u21a9
Cibulskis, K., et al., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol, 2013. 31(3): p. 213-9.\u00a0\u21a9
Benjamin, D., et al., Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 2019: p. 861054.\u00a0\u21a9
Kim, S., et al., Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods, 2018. 15(8): p. 591-594.\u00a0\u21a9
Lai, Z., et al., VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res, 2016. 44(11): p. e108.\u00a0\u21a9
McLaren, W., et al., The Ensembl Variant Effect Predictor. Genome Biol, 2016. 17(1): p. 122.\u00a0\u21a9
Memorial Sloan Kettering Cancer Center. vcf2maf. 2013; Available from: https://github.com/mskcc/vcf2maf.\u00a0\u21a9
Boeva, V., et al., Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics, 2012. 28(3): p. 423-5.\u00a0\u21a9
Favero, F., et al., Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol, 2015. 26(1): p. 64-70.\u00a0\u21a9
Pedersen, B. somalier: extract informative sites, evaluate relatedness, and perform quality-control on BAM/CRAM/BCF/VCF/GVCF. 2018; Available from: https://github.com/brentp/somalier.\u00a0\u21a9
Wood, D.E., J. Lu, and B. Langmead, Improved metagenomic analysis with Kraken 2. Genome Biol, 2019. 20(1): p. 257.\u00a0\u21a9
Wingett, S.W. and S. Andrews, FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res, 2018. 7: p. 1338.\u00a0\u21a9
Okonechnikov, K., A. Conesa, and F. Garcia-Alcalde, Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics, 2016. 32(2): p. 292-4.\u00a0\u21a9
Cingolani, P., et al., A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 2012. 6(2): p. 80-92.\u00a0\u21a9
Ewels, P., et al., MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 2016. 32(19): p. 3047-8.\u00a0\u21a9
Koster, J. and S. Rahmann, Snakemake-a scalable bioinformatics workflow engine. Bioinformatics, 2018. 34(20): p. 3600.\u00a0\u21a9
The output files and their locations are broken down here for the XAVIER pipeline. Pre-processing and germline variant calling steps are common but somatic variant calling is dependent on whether the pipeline was run in either (A) tumor-normal pair or (B) tumor-only analysis mode. All file locations are relative to the output directory specified during the job submission.
The output directory after a complete XAVIER run should look like:
xavier_output/\n\u251c\u2500\u2500 bams\n\u251c\u2500\u2500 cluster.json # cluster info for the run\n\u251c\u2500\u2500 config\n\u251c\u2500\u2500 config.json # config file for the run\n\u251c\u2500\u2500 fastqs\n\u251c\u2500\u2500 germline\n\u251c\u2500\u2500 indels.vcf.gz[.tbi] # raw germline INDELs\n\u251c\u2500\u2500 input_files\n\u251c\u2500\u2500 intervals.list\n\u251c\u2500\u2500 {sample1}-normal.R1.fastq.gz -> /path/to/{sample1}-normal.R1.fastq.gz\n\u251c\u2500\u2500 {sample1}-normal.R2.fastq.gz -> /path/to/{sample1}-normal.R2.fastq.gz\n\u251c\u2500\u2500 {sample1}-tumor.R1.fastq.gz -> /path/to/{sample1}-tumor.R1.fastq.gz\n\u251c\u2500\u2500 {sample1}-tumor.R2.fastq.gz -> /path/to/{sample1}-tumor.R2.fastq.gz\n.\n.\n.\n\u251c\u2500\u2500 kickoff.sh\n\u251c\u2500\u2500 logfiles\n\u251c\u2500\u2500 QC\n\u251c\u2500\u2500 resources\n\u251c\u2500\u2500 snps.vcf.gz[.tbi] # raw germline SNPs\n\u251c\u2500\u2500 somatic_paired # in case of tumor-normal paired run\n\u251c\u2500\u2500 somatic_tumor_only # in case of tumor-only run\n\u2514\u2500\u2500 workflow\n
Below we describe the different folders that contain specific outputs obtained for all samples from the XAVIER pipeline
The QC folder contains all the Quality-Control analyses performed at different steps of the pipeline for each sample to assess sequencing quality before and after adapter trimming, microbial taxonomic composition, contamination, variant calling, etc. The final summary report and data is available finalQC folder. \\ The MultiQC report also contains results from other analysis like mapping statistics, ancestry and relatedness, etc. It is recommended to study the MultiQC report first to get a birds eye view of the sequence data quality.
The bams folder contain two subfolders chrom_split and final_bams. final_bams contains the final processed BAM files for each sample in the run and the chrom_split folder contains all the sample BAM files split by each chromosome.
This folder contains the output from the GATK Best Practices pipeline to obtain germline variants with a few alterations detailed below. Briefly, joint SNP and INDEL variant detection is conducted across all samples included in a pipeline run using the GATK Haplotypcaller under default settings. Raw variants are then subsequently filtered based on several GATK annotations: \\ A strict set of criteria (QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum < -12.5, ReadPosRankSum < -8.0 for SNPs; QD < 2.0, FS > 200.0, ReadPosRankSum < -20.0 for INDELs) generates the 'combined.strictFilter.vcf'.
This call set is highly stringent, maximizing the true positive rate at the expense of an elevated false negative rate. This call set is really only intended for more general population genetic scale analyses (e.g., burden tests, admixture, linkage/pedigree based analysis, etc.) where false positives can be significantly confounding.
In case of human sequence data, a basic analyses of sample relatedness and ancestry (e.g., % European, African, etc.) is also performed using somalier.
The output folder looks like:
germline/\n\u251c\u2500\u2500 gVCFs\n.\n.\n.\n\u251c\u2500\u2500 somalier # only for hg38 genome\n\u2514\u2500\u2500 VCF\n
The VCF folder contains the final filtered germline variants (SNPs and INDELs) for all samples combined. The folder also contains raw variants for each sample, all samples combined, and also combined raw variants split by chromosome.
This folder contains the snakemake log files and computational statistics for the XAVIER run. All the log files (i.e., standard output and error) for each individual step are in the slurmfiles folder. These logfiles are important to diagnose errors in case the pipeline fails.
This workflow calls somatic SNPs and INDELs using multiple variant detection algorithms. For each of these tools, variants are called in a paired tumor-normal fashion, with default settings. See Pipeline Details for more information about the tools used and their parameter settings.
For each sample, the resulting VCF is fully annotated using VEP and converted to a MAF file using the vcf2maf tool. Resulting MAF files are found in maf folder within each caller's results directory (i.e., mutect2_out, strelka_out, etc.). Individual sample MAF files are then merged and saved in merged_somatic_variants directory.
For Mutect2, we use a panel of normals (PON) developed from the ExAC (excluding TCGA) dataset, filtered for variants <0.001 in the general population, and also including and in-house set of blacklisted recurrent germline variants that are not found in any population databases.
For Copy Number Variants (CNVs), two tools are employed in tandem. First, Control-FREEC is run with default parameters. This generates pileup files that can be used by Sequenza, primarily for jointly estimating contamination and ploidy. These value are used to run Freec a second time for improved performance.
In general, the tumor-only pipeline is a stripped down version of the tumor-normal pipeline. We only run MuTect2, Mutect, and VarDict for somatic variant detection, with the same PON and filtering as described above for the tumor-normal pipeline.
Workflow diagram of the XAVIER: the pipeline is composed of a series of data processing steps to trim, align, and recalibrate reads prior to calling variants. These data processing steps closely follow GATK's best pratices for cleaning up raw alignments. The pipeline also consists of a series of comprehensive quality-control steps.
Removes variants with certain flags from vardict; (1) Germline: detected in germline sample (pass all quality parameters); (2) LikelyLOH: detected in germline but either lost in tumor OR 20-80% in germline, but increased to 1-opt_V (95%); (3) AFDiff: detected in tumor (pass quality parameters) and present in germline but didn\u2019t pass quality parameters.
varscan
pileup
-d 100000 -q 15 -Q 15
samtools mpileup arguments; max depth of 100,000; min mapping quality of 15; min base quality of 15
calling
--strand-filter 0
Do not ignore variants with >90% support on one strand
--min-var-freq 0.01
Minimum variant allele frequency threshold 0.01
--output-vcf 1
Output in VCF format
--variants 1
Report only variant (SNP/indel) positions
all
GATK SelectVariants
--exclude-filtered
Removes non-PASS variants
--discordance
Remove variants found in supplied file (same as panel-of-normals file)
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier cache sub command in more detail. With minimal configuration, the cache sub command enables you to cache remote resources for the xavier pipeline. Caching remote resources allows the pipeline to run in an offline mode. The cache sub command can also be used to pull our pre-built reference bundles onto a new cluster or target system.
The cache sub command creates local cache on the filesysytem for resources hosted on DockerHub or AWS S3. These resources are normally pulled onto the filesystem when the pipeline runs; however, due to network issues or DockerHub pull rate limits, it may make sense to pull the resources once so a shared cache can be created and re-used. It is worth noting that a singularity cache cannot normally be shared across users. Singularity strictly enforces that its cache is owned by the user. To get around this issue, the cache subcommand can be used to create local SIFs on the filesystem from images on DockerHub.
XAVIER pipeline can be executed from either using the graphical user interface (GUI) or the command line interface (CLI). GUI offers a more interactive way for the user to provide input and adjust parameter settings. This part of the documentation describes how to run xavier using the GUI (with screenshots). See Command Line tab to read more about the xavier executable and running XAVIER pipeline using the CLI.
"},{"location":"usage/gui/#2-setting-up-xavier","title":"2. Setting up XAVIER","text":""},{"location":"usage/gui/#21-login-to-cluster","title":"2.1 Login to cluster","text":"
# Setup Step 1.) ssh into cluster's head node\n# example below for Biowulf cluster\nssh -Y $USER@biowulf.nih.gov\n
"},{"location":"usage/gui/#22-grab-an-interactive-node","title":"2.2 Grab an interactive node","text":"
# Setup Step 2.) Please do not run XAVIER on the head node!\n# Grab an interactive node first\nsinteractive --time=12:00:00 --mem=8gb --cpus-per-task=4\n
NOTE: ccbrpipeliner is a custom module created on biowulf which contains various NGS data analysis pipelines developed, tested, and benchmarked by experts at CCBR.
# Setup Step 3.) Add ccbrpipeliner module\nmodule purge # to reset the module environment\nmodule load ccbrpipeliner\n
If the module was loaded correctly, the greetings message should be displayed.
[+] Loading ccbrpipeliner 5 ...\n###########################################################################\n CCBR Pipeliner\n###########################################################################\n \"ccbrpipeliner\" is a suite of end-to-end pipelines and tools\n Visit https://github.com/ccbr for more details.\n Pipelines are available on BIOWULF and FRCE.\n Tools are available on BIOWULF, HELIX and FRCE.\n\n The following pipelines/tools will be loaded in this module:\n\n RENEE v2.5 https://ccbr.github.io/RENEE/\n XAVIER v3.0 https://ccbr.github.io/XAVIER/\n CARLISLE v2.4 https://ccbr.github.io/CARLISLE/\n CHAMPAGNE v0.2 https://ccbr.github.io/CHAMPAGNE/\n CRUISE v0.1 https://ccbr.github.io/CRUISE/\n\n spacesavers2 v0.10 https://ccbr.github.io/spacesavers2/\n permfix v0.6 https://github.com/ccbr/permfix\n###########################################################################\nThank you for using CCBR Pipeliner\n###########################################################################\n
To run the XAVIER pipeline from the GUI, simply enter:
xavier gui\n
and it will launch the XAVIER window.
Note: Please wait until window created! message appears on the terminal.
"},{"location":"usage/gui/#32-folder-paths-and-reference-genomes","title":"3.2 Folder paths and reference genomes","text":"
To enter the location of the input folder containing FASTQ files and the location where the output folders should be created, either simply type the absolute paths
or use the Browse tab to choose the input and output directories
Next, from the drop down menu select the reference genome (hg38/mm10)
In case the paired normal samples are unavailable, XAVIER pipeline can be run in tumor-only mode which does not require paired samples information. However, in the absence of matching normal samples, CNV analysis is also unavailable.
After all the information is filled out, press Submit.
If the pipeline detects no errors and the run was submitted, a new window appears that has the output of a \"dry-run\" which summarizes each step of the pipeline.
Click OK
A dialogue box will popup to confirm submitting the job to slurm.
Click Yes
The dry-run output will be displayed again and the master job will be submitted. An email notification will be sent out when the pipeline starts and ends.
The XAVIER gui will ask to submit another job.
Click Yes to start again or No to close the XAVIER gui.
Users can input certain additional settings for the pipeline run including running an additional step to correct strand orientation bias in Formalin-Fixed Paraffin-Embedded (FFPE) samples and to provide a custom exome targets BED file. This file can be obtained from the manufacturer of the target capture kit that was used.
"},{"location":"usage/gui/#4-special-instructions-for-biowulf","title":"4. Special instructions for Biowulf","text":"
XAVIER GUI natively uses the X11 Window System to run XAVIER pipeline and display the graphics on a personal desktop or laptop. However, if running XAVIER specifically on NIH's Biowulf cluster, the HPC staff recommends NoMachine (NX) to run graphics applications.
Please see details here on how to install and connect to Biowulf on your local computer using NoMachine.
Once connected to Biowulf using NX, right click to open a terminal connection
and start an interactive session.
Similar to the instructions above, load ccbrpipeliner module and enter xavier gui to launch the XAVIER gui.
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier run sub command in more detail. With minimal configuration, the run sub command enables you to start running xavier pipeline.
Setting up the xavier pipeline is fast and easy! In its most basic form, xavier run only has four required inputs.
The synopsis for each command shows its parameters and their usage. Optional parameters are shown in square brackets.
A user must provide a list of FastQ or BAM files (globbing is supported) to analyze via --input argument, an output directory to store results via --output argument, an exome targets BED file for the samples' capture kit, and select reference genome for alignment and annotation via the --genome argument.
Use you can always use the -h option for information on a specific command.
Each of the following arguments are required. Failure to provide a required argument will result in a non-zero exit-code.
--input INPUT [INPUT ...]
Input FastQ or BAM file(s) to process. type: file(s)
One or more FastQ files can be provided. The pipeline does NOT support single-end WES data. Please provide either a set of FastQ files or a set of BAM files. The pipeline does NOT support processing a mixture of FastQ files and BAM files. From the command-line, each input file should separated by a space. Globbing is supported! This makes selecting FastQ files easy. Input FastQ files should be gzipp-ed.
This location is where the pipeline will create all of its output files, also known as the pipeline's working directory. If the provided output directory does not exist, it will be initialized automatically.
Example: --output /data/$USER/WES_hg38
--runmode {init,dryrun,run} `
Execution Process. type: string
User should initialize the pipeline folder by first running --runmode init User should then perform a dry-run to list all steps the pipeline will take--runmode dryrun User should then perform the full run --runmode run
Example: --runmode init THEN --runmode dryrun THEN --runmode run
--genome {hg38, custom.json}
Reference genome. type: string/file
This option defines the reference genome for your set of samples. On Biowulf, xavier does comes bundled with pre built reference files for human samples; however, it is worth noting that the pipeline does accept a pre-built resource bundle pulled with the cache sub command (coming soon). Currently, the pipeline only supports the human reference hg38; however, support for mouse reference mm10 will be added soon.
Pre built Option Here is a list of available pre built genomes on Biowulf: hg38, mm10.
Custom Option For users running the pipeline outside of Biowulf, a pre-built resource bundle can be pulled with the cache sub command (coming soon). Please supply the custom reference JSON file that was generated by the cache sub command.
Example: --genome hg38 OR --genome /data/${USER}/hg38/hg38.json
--targets TARGETS
Exome targets BED file. type: file
This file can be obtained from the manufacturer of the target capture kit that was used.
If not provided, the default targets file from the genome config is used
Each of the following arguments are optional and do not need to be provided.
-h, --help
Display Help. type: boolean flag
Shows command's synopsis, help message, and an example command
Example: --help
--silent
Silence standard output. type: boolean flag
Reduces the amount of information directed to standard output when submitting master job to the job scheduler. Only the job id of the master job is returned.
Example: --silent
--mode {local,slurm}
Execution Method. type: string default: slurm
Execution Method. Defines the mode or method of execution. Valid mode options include: local or slurm.
local Local executions will run serially on compute instance. This is useful for testing, debugging, or when a users does not have access to a high performance computing environment. If this option is not provided, it will default to a local execution mode.
slurm The slurm execution method will submit jobs to a cluster using a singularity backend. It is recommended running xavier in this mode as execution will be significantly faster in a distributed environment.
Example: --mode slurm
--job-name JOB_NAME
Set the name of the pipeline's master job. type: string > default: pl:xavier
When submitting the pipeline to a job scheduler, like SLURM, this option always you to set the name of the pipeline's master job. By default, the name of the pipeline's master job is set to \"pl:xavier\".
List of variant callers to detect mutations. Please select from one or more of the following options: [mutect2, mutect, strelka, vardict, varscan]. Defaults to using all variant callers.
Example: --callers mutect2 strelka varscan
--pairs PAIRS
Tumor normal pairs file. type: file
This tab delimited file contains two columns with the names of tumor and normal pairs, one per line. The header of the file needs to be Tumor for the tumor column and Normal for the normal column. The base name of each sample should be listed in the pairs file. The base name of a given sample can be determined by removing the following extension from the sample's R1 FastQ file: .R1.fastq.gz. Contents of example pairs file:
Normal Tumor\nSample4_CRL1622_S31 Sample10_ARK1_S37\nSample4_CRL1622_S31 Sample11_ACI_158_S38\n
Example: --pairs /data/$USER/pairs.tsv
--ffpe
Apply FFPE correction. type: boolean flag
Runs an additional steps to correct strand orientation bias in Formalin-Fixed Paraffin-Embedded (FFPE) samples. Do NOT use this option with non-FFPE samples.
Example: --ffpe
--cnv
Call copy number variations (CNVs). type: boolean flag
CNVs will only be called from tumor-normal pairs. If this option is provided without providing a --pairs file, CNVs will NOT be called.
Example: --cnv
--singularity-cache SINGULARITY_CACHE
Overrides the $SINGULARITY_CACHEDIR environment variable. type: path default: --output OUTPUT/.singularity
Singularity will cache image layers pulled from remote registries. This ultimately speeds up the process of pull an image from DockerHub if an image layer already exists in the singularity cache directory. By default, the cache is set to the value provided to the --output argument. Please note that this cache cannot be shared across users. Singularity strictly enforces you own the cache directory and will return a non-zero exit code if you do not own the cache directory! See the --sif-cache option to create a shareable resource.
Path where a local cache of SIFs are stored. type: path
Uses a local cache of SIFs on the filesystem. This SIF cache can be shared across users if permissions are set correctly. If a SIF does not exist in the SIF cache, the image will be pulled from Dockerhub and a warning message will be displayed. The xavier cache subcommand can be used to create a local SIF cache. Please see xavier cache for more information. This command is extremely useful for avoiding DockerHub pull rate limits. It also remove any potential errors that could occur due to network issues or DockerHub being temporarily unavailable. We recommend running xavier with this option when ever possible.
Example: --singularity-cache /data/$USER/SIFs
--threads THREADS
Max number of threads for each process. type: int default: 2
Max number of threads for each process. This option is more applicable when running the pipeline with --mode local. It is recommended setting this value to the maximum number of CPUs available on the host machine.
# Step 1.) Grab an interactive node\n# Do not run on head node!\nsinteractive --mem=8g --cpus-per-task=4\nmodule purge\nmodule load ccbrpipeliner\n\n# Step 2A.) Initialize the all resources to the output folder\nxavier run --input tests/data/*.R?.fastq.gz \\\n --output /data/$USER/xavier_hg38 \\\n --genome hg38 \\\n --targets Agilent_SSv7_allExons_hg38.bed \\\n --mode slurm \\\n --runmode init\n\n# Step 2B.) Dry-run the pipeline\nxavier run --input tests/data/*.R?.fastq.gz \\\n --output /data/$USER/xavier_hg38 \\\n --genome hg38 \\\n --targets Agilent_SSv7_allExons_hg38.bed \\\n --mode slurm \\\n --runmode dryrun\n\n# Step 2C.) Run the XAVIER pipeline\n# The slurm mode will submit jobs to the cluster.\n# It is recommended running xavier in this mode.\nxavier run --input tests/data/*.R?.fastq.gz \\\n --output /data/$USER/xavier_hg38 \\\n --genome hg38 \\\n --targets Agilent_SSv7_allExons_hg38.bed \\\n --mode slurm \\\n --runmode run\n
The example dataset in tests/data in this repository is a very small subsampled dataset, and some steps of the pipeline fail due to the small size (CNV callling, somalier, etc). We have a larger subsample (25% of a full human dataset) available on Biowulf if you would like to test the full functionality of the pipeline: /data/CCBR_Pipeliner/testdata/XAVIER/human_subset/*.R?.fastq.gz
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier unlock sub command in more detail. With minimal configuration, the unlock sub command enables you to unlock a pipeline output directory.
If the pipeline fails ungracefully, it maybe required to unlock the working directory before proceeding again. Snakemake will inform a user when it maybe necessary to unlock a working directory with an error message stating: Error: Directory cannot be locked.
Please verify that the pipeline is not running before running this command. If the pipeline is currently running, the workflow manager will report the working directory is locked. The is the default behavior of snakemake, and it is normal. Do NOT run this command if the pipeline is still running! Please kill the master job and it's child jobs prior to running this command.
Unlocking xavier pipeline output directory is fast and easy! In its most basic form, xavier unlock only has one required input.
The synopsis for this command shows its parameters and their usage. Optional parameters are shown in square brackets.
A user must provide an output directory to unlock via --output argument. After running the unlock sub command, you can resume the build or run pipeline from where it left off by re-running it.
Use you can always use the -h option for information on a specific command.
Path to a previous run's output directory. This will remove a lock on the working directory. Please verify that the pipeline is not running before running this command. Example: --output /data/$USER/WES_hg38
# Step 0.) Grab an interactive node (do not run on head node)\nsinteractive --mem=8g -N 1 -n 4\nmodule purge\nmodule load ccbrpipeliner\n\n# Step 1.) Unlock a pipeline output directory\nxavier unlock --output /data/$USER/xavier_hg38\n
"}]}
\ No newline at end of file
diff --git a/dev/sitemap.xml.gz b/dev/sitemap.xml.gz
index b75efa86c7320bab7fdb8b8e7afd907128bb3b86..9ae7875e8550fd047e52e5d586b379c332aaaea0 100644
GIT binary patch
delta 13
Ucmb=gXP58h;8>jeW+Hn903Q4V!Txavier cache - XAVIER Documentation
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier cache sub command in more detail. With minimal configuration, the cache sub command enables you to cache remote resources for the xavier pipeline. Caching remote resources allows the pipeline to run in an offline mode. The cache sub command can also be used to pull our pre-built reference bundles onto a new cluster or target system.
The cache sub command creates local cache on the filesysytem for resources hosted on DockerHub or AWS S3. These resources are normally pulled onto the filesystem when the pipeline runs; however, due to network issues or DockerHub pull rate limits, it may make sense to pull the resources once so a shared cache can be created and re-used. It is worth noting that a singularity cache cannot normally be shared across users. Singularity strictly enforces that its cache is owned by the user. To get around this issue, the cache subcommand can be used to create local SIFs on the filesystem from images on DockerHub.
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier cache sub command in more detail. With minimal configuration, the cache sub command enables you to cache remote resources for the xavier pipeline. Caching remote resources allows the pipeline to run in an offline mode. The cache sub command can also be used to pull our pre-built reference bundles onto a new cluster or target system.
The cache sub command creates local cache on the filesysytem for resources hosted on DockerHub or AWS S3. These resources are normally pulled onto the filesystem when the pipeline runs; however, due to network issues or DockerHub pull rate limits, it may make sense to pull the resources once so a shared cache can be created and re-used. It is worth noting that a singularity cache cannot normally be shared across users. Singularity strictly enforces that its cache is owned by the user. To get around this issue, the cache subcommand can be used to create local SIFs on the filesystem from images on DockerHub.
XAVIER pipeline can be executed from either using the graphical user interface (GUI) or the command line interface (CLI). GUI offers a more interactive way for the user to provide input and adjust parameter settings. This part of the documentation describes how to run xavier using the GUI (with screenshots). See Command Line tab to read more about the xavier executable and running XAVIER pipeline using the CLI.
XAVIER pipeline can be executed from either using the graphical user interface (GUI) or the command line interface (CLI). GUI offers a more interactive way for the user to provide input and adjust parameter settings. This part of the documentation describes how to run xavier using the GUI (with screenshots). See Command Line tab to read more about the xavier executable and running XAVIER pipeline using the CLI.
# Setup Step 2.) Please do not run XAVIER on the head node!
diff --git a/dev/usage/run/index.html b/dev/usage/run/index.html
index 54790de..8062a4a 100644
--- a/dev/usage/run/index.html
+++ b/dev/usage/run/index.html
@@ -1,4 +1,4 @@
- xavier run - XAVIER Documentation
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier run sub command in more detail. With minimal configuration, the run sub command enables you to start running xavier pipeline.
Setting up the xavier pipeline is fast and easy! In its most basic form, xavier run only has four required inputs.
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier run sub command in more detail. With minimal configuration, the run sub command enables you to start running xavier pipeline.
Setting up the xavier pipeline is fast and easy! In its most basic form, xavier run only has four required inputs.
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier unlock sub command in more detail. With minimal configuration, the unlock sub command enables you to unlock a pipeline output directory.
If the pipeline fails ungracefully, it maybe required to unlock the working directory before proceeding again. Snakemake will inform a user when it maybe necessary to unlock a working directory with an error message stating: Error: Directory cannot be locked.
Please verify that the pipeline is not running before running this command. If the pipeline is currently running, the workflow manager will report the working directory is locked. The is the default behavior of snakemake, and it is normal. Do NOT run this command if the pipeline is still running! Please kill the master job and it's child jobs prior to running this command.
Unlocking xavier pipeline output directory is fast and easy! In its most basic form, xavier unlock only has one required input.
The xavier executable is composed of several inter-related sub commands. Please see xavier -h for all available options.
This part of the documentation describes options and concepts for xavier unlock sub command in more detail. With minimal configuration, the unlock sub command enables you to unlock a pipeline output directory.
If the pipeline fails ungracefully, it maybe required to unlock the working directory before proceeding again. Snakemake will inform a user when it maybe necessary to unlock a working directory with an error message stating: Error: Directory cannot be locked.
Please verify that the pipeline is not running before running this command. If the pipeline is currently running, the workflow manager will report the working directory is locked. The is the default behavior of snakemake, and it is normal. Do NOT run this command if the pipeline is still running! Please kill the master job and it's child jobs prior to running this command.
Unlocking xavier pipeline output directory is fast and easy! In its most basic form, xavier unlock only has one required input.
The synopsis for this command shows its parameters and their usage. Optional parameters are shown in square brackets.
A user must provide an output directory to unlock via --output argument. After running the unlock sub command, you can resume the build or run pipeline from where it left off by re-running it.
Use you can always use the -h option for information on a specific command.
Path to a previous run's output directory. This will remove a lock on the working directory. Please verify that the pipeline is not running before running this command. Example:--output /data/$USER/WES_hg38