![]()

Snakemake pipeline for microbiome diversity effect size benchmarking
NOTE: Please note that xebec is still under active development.
To use xebec, you will need several dependencies.
We recommend using mamba to install these packages when possible.
mamba install -c conda-forge -c bioconda biom-format h5py==3.1.0 scipy==1.8 numpy==1.23 snakemake pandas unifrac scikit-bio bokeh==3.1.0 unifrac-binaries jinja2
pip install evident>=0.4.0 gemelli>=0.0.8
To install xebec, run the following command from the command line:
pip install xebec
If you run xebec --help, you should see the following:
$ xebec --help
Usage: xebec [OPTIONS]
Options:
--version Show the version and exit.
-ft, --feature-table PATH Feature table in BIOM format. [required]
-m, --metadata PATH Sample metadata in TSV format. [required]
-t, --tree PATH Phylogenetic tree in Newick format.
[required]
-o, --output PATH Output workflow directory. [required]
--max-category-levels INTEGER Max number of levels in a category.
[default: 5]
--min-level-count INTEGER Min number of samples per level per
category. [default: 3]
--rarefy-percentile FLOAT Percentile of sample depths at which to
rarefy. [default: 10]
--n-pcoa-components INTEGER Number of PCoA components to compuate.
[default: 3]
--validate-input / --no-validate-input
Whether to validate input before creating
workflow. [default: validate-input]
--help Show this message and exit.
To create the workflow structure, pass in the filepaths for the feature table, sample metadata, and phylogenetic tree. You must also pass in a path to a directory in which to create the workflow. Additionally, you can provide parameters for determining how to process your sample metadata.
After running this command, navigate inside the output directory you created.
There should be two subdirectories: workflow/ and config/.
To start the pipeline , run the following command:
snakemake --cores 1
You should see the Snakemake pipeline start running the jobs.
If this pipeline runs sucessfully, the processed results will be located at results/.
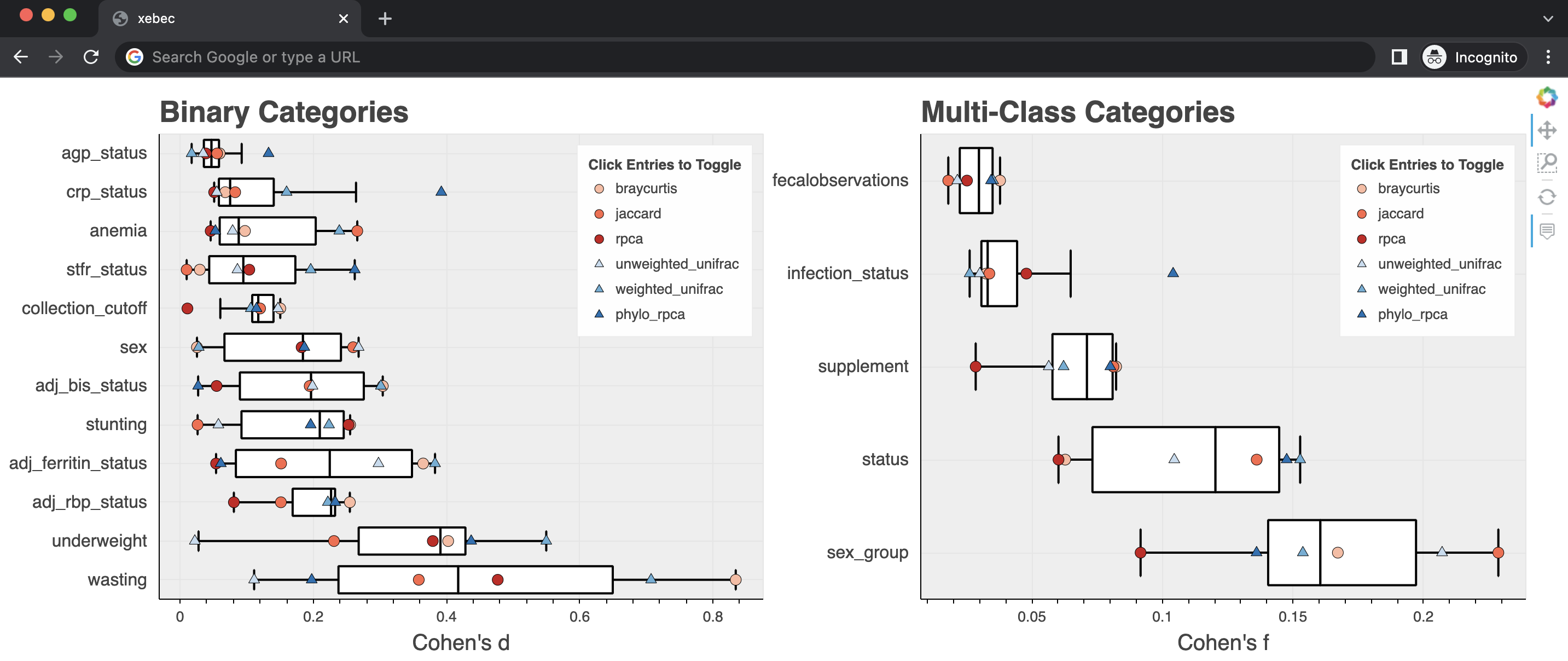
Included in the results are the concatenated effect size values as well as interactive plots summarizing the effect sizes for each metadata column for each diversity metric.
These plots are generated using Bokeh and can be visualized in any modern web browser.
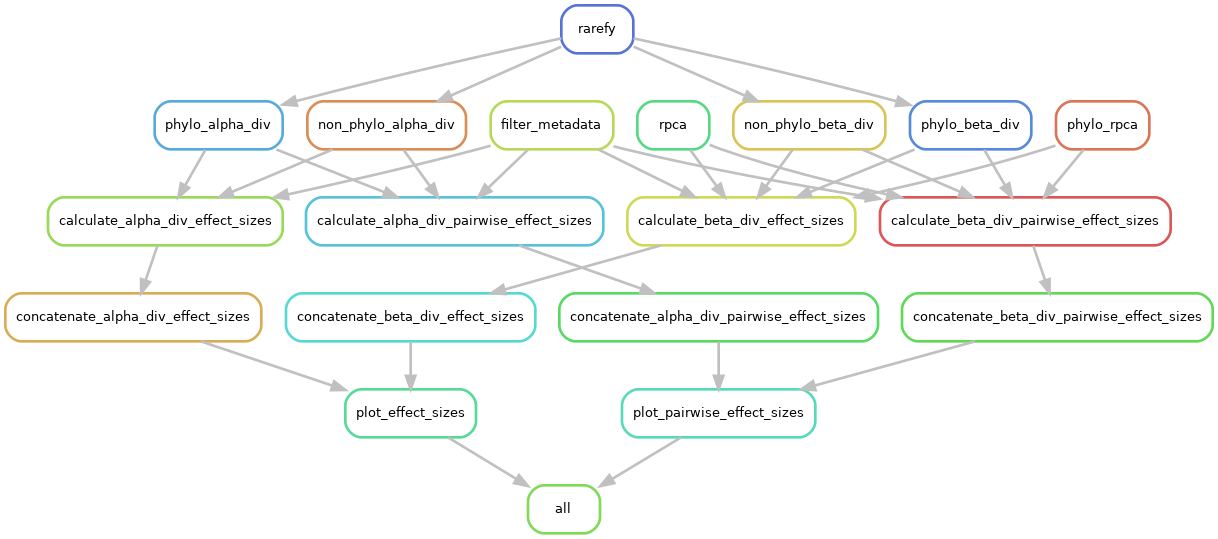
xebec performs four main steps, some of which have substeps.
- Process data (filter metadata, rarefaction)
- Run diversity analyses
- Calculate effect sizes (concatenate together)
- Generate visualizations
An overview of the DAG is shown below:
xebec allows configuration of what alpha and beta diversity metrics are included in the workflow.
To add or remove metrics, modify the config/alpha_div_metrics.yml and config/beta_div_metrics.yml files.
For alpha diversity, any metric that can be passed into skbio.alpha_diversity should work.
For beta diversity, any non-phylogenetic metric that can be passed into skbio.beta_diversity should work.
Valid phylogenetic beta diversity are those that can be passed into Striped UniFrac.
Make sure that any additional diversity metrics are annotated with phylo or non_phylo so xebec knows how to process them.
The xebec workflow can be decorated with many configuration options available in Snakemake, including resource usage and HPC scheduling. We recommend reading through the Snakemake documentation for details on these options. Note that some of these options may require creating new configuration files.
If you have any issues with xebec, please leave a GitHub issue or pull request.