| jupyter | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Ludwig Schneider, Pablo Zubieta, and Juan de Pablo
Pritzker School of Molecular Engineering
The University of Chicago
First, we set up our environment. We will be using a pre-compiled and packaged installation of HOOMD-blue and the hoomd-dlext plugin. It will be downloaded from Google Drive and made accessible to the Python process running in this Colab instance.
BASE_URL="https://drive.usercontent.google.com/download?id=1hsKkKtdxZTVfHKgqVF6qV2e-4SShmhr7"
COOKIES="/tmp/cookies.txt"
CONFIRMATION="$(wget -q --save-cookies $COOKIES --keep-session-cookies --no-check-certificate $BASE_URL -O- | sed -rn 's/.*confirm=(\w+).*/\1\n/p')"
wget -q --load-cookies $COOKIES "$BASE_URL&confirm=$CONFIRMATION" -O pysages-env.zip
rm -rf $COOKIES%env PYSAGES_ENV=/env/pysagesmkdir -p $PYSAGES_ENV .
unzip -qquo pysages-env.zip -d $PYSAGES_ENVimport os
import sys
ver = sys.version_info
sys.path.append(os.environ["PYSAGES_ENV"] + "/lib/python" + str(ver.major) + "." + str(ver.minor) + "/site-packages/")
os.environ["LD_LIBRARY_PATH"] = "/usr/lib/x86_64-linux-gnu:" + os.environ["LD_LIBRARY_PATH"]We'll also need some additional python dependencies
!pip install -qq "numpy<2" gsd > /dev/nullThe next step is to install PySAGES. First, we need to install JAX. Fortunately, Colab already ships with JAX pre-installed (to learn how to install it you can look at the JAX documentation for more details). To install PySAGES, we retrieve the latest version from GitHub and add its dependencies via pip.
!pip install -qq git+https://github.com/SSAGESLabs/PySAGES.git > /dev/nullmkdir /content/advanced_sampling
cd /content/advanced_samplingMany systems have rugged free-energy landscapes, where different basins are divided by energy barriers.
- Protein folding
- Protein-ligand binding
- Liquid membrane fusion


- Liquid-vapor transition
- Crystallization
- Shape transitions
- Magnetization (Ising model)
Wikipedia: https://en.wikipedia.org/wiki/Ising_model
Also known as order parameters in other contexts.
Usually, such transitions can be described in terms of order parameters/collective variables (CV).
Examples:
- center of mass of the molecule
- opening angle of protein
- magnetization
- more common options in PySAGES
Let's start exploring an easy system and find the free-energy landscape.
- a priori
$A(\xi)$ is unknown - but we can measure the density of states
$p(\xi)$
We can recover the free-energy profile from
-
$A$ : free-energy -
$Z$ : NVT partition function
We can formulate the partial partition function for a collective variable
With regular MC/MD a finite simulation trajectory is confined to a single basin as the probability of leaving the basin scales as
- system might not equilibrate
- trajectory is not ergodic
Let's examine such a system via computer simulations.
- fast to integrate
- custom bond potential to shape the free-energy landscape
import numpy as np
def energy_and_forces(x, amplitude=1., roughness=5, periodicity=1):
omega = np.pi * periodicity
energy = x**2
energy += (1 - np.exp(-x**2)) * roughness * np.cos(omega * x)
energy *= amplitude
forces = 2 * x
forces -= omega * roughness * (1 - np.exp(-x**2)) * np.sin(omega * x)
forces += 2 * roughness * np.exp(-x**2) * x * np.cos(omega * x)
forces *= -amplitude
return energy, forces
def energy(x, **kwargs):
return energy_and_forces(x, **kwargs)[0]
def forces(x, **kwargs):
return energy_and_forces(x, **kwargs)[1]- customizable roughness and steepness
- symmetric around the origin
import matplotlib.pyplot as plt
fig, ax = plt.subplots()
ax.set_xlabel(r"$r$ $[\sigma]$")
ax.set_ylabel(r"$E$ $[k_B T]$")
ax.set_xlim((0,4))
x = np.linspace(0,4,100)
ax.plot(x, energy(x), label="reference")
ax.plot(x, energy(x, roughness=9), label="rougher")
ax.plot(x, energy(x, amplitude=2), label="steeper")
ax.plot(x, energy(x, periodicity=2), label="more minima")
# Uncomment to inspect the forces
# ax.plot(x, forces(x), label="analytic force")
# ax.plot(x[:-1], -np.diff(energy(x)) / (x[1] - x[0]), label="numeric force")
ax.legend(loc="best")
fig.show()- We can choose and adjust the potential
Here we choose a system with multiple basins separated by energy barriers.
However, the free energy of this system is not the energy of this bond.
Because we are using a distance the system is best described in radial coordinates, which includes the Jacobi determinant
Hence we can obtain the free energy with a logarithmic correction.
import matplotlib.pyplot as plt
fig, ax = plt.subplots()
ax.set_xlabel(r"$r$ $[\sigma]$")
ax.set_ylabel(r"$A$ $[k_B T]$")
ax.set_xlim((0,4))
def free_energy(energy, kT: float = 1):
"""
Modifies function energy, such that it returns the free energy
with the appropriate logarithmic correction.
"""
beta = 1 / kT
tau = 2 * np.pi
def log_corrected_energy(x):
corrected_energy = beta * energy(x) - np.log(tau * x**2)
corrected_energy -= corrected_energy[0]
return corrected_energy
return log_corrected_energy
x = np.linspace(0.01, 4, 100)
ax.plot(x, free_energy(energy)(x), label="reference")
ax.plot(x, free_energy(lambda x: energy(x, roughness=9))(x), label="rougher")
ax.plot(x, free_energy(lambda x: energy(x, amplitude=2))(x), label="steeper")
ax.plot(x, free_energy(lambda x: energy(x, periodicity=2))(x), label="more minima")
ax.legend(loc="best")
fig.show()- Notice how the unlikely short distances remove the first minimum around 0
- use of a single bond between two particles to realize the potential in HOOMD-blue
import hoomd
import gsd.hoomd
kT = 1
dt = 1e-3
fes_params = dict(amplitude=1, roughness=5, periodicity=1)
def generate_context(kT=kT, dt=dt, fes_params=fes_params, **kwargs):
"""
Generates a simulation context, we pass this function to the attribute
`run` of our sampling method.
"""
sim = hoomd.Simulation(device=hoomd.device.auto_select(), seed=42)
# System Definition
snapshot = gsd.hoomd.Frame()
snapshot.configuration.box = [50, 50, 50, 0, 0, 0]
snapshot.particles.N = 2
snapshot.particles.types = ['P', 'G']
snapshot.particles.typeid = [0, 1]
snapshot.particles.position = [[3.0, 0, 0], [0, 0, 0]]
snapshot.bonds.N = 1
snapshot.bonds.types = ["bond"]
snapshot.bonds.typeid = [0]
snapshot.bonds.group = [[0, 1]]
sim.create_state_from_snapshot(snapshot)
sim.run(0)
integrator = hoomd.md.Integrator(dt=dt)
# Interaction Potential
r_min, r_max = 0, 10
n_points = 512
fes_points = np.linspace(r_min, r_max, n_points)
energy, forces = energy_and_forces(fes_points, **fes_params)
fes = hoomd.md.bond.Table(n_points)
fes.params["bond"] = dict(r_min=r_min, r_max=r_max, U=energy, F=forces)
integrator.forces.append(fes)
mobile_particles = hoomd.filter.Type("P")
langevin = hoomd.md.methods.Langevin(filter=mobile_particles, kT=kT)
integrator.methods.append(langevin)
sim.operations.integrator = integrator
return simThe bond realizes the custom energy landscape between two particles. In this case, we can determine the free energy with the Jacobian correction and the bond energy. This is only true because of the simple nature of this toy system. Usually, the free energy is unknown and needs to be discovered via advanced sampling methods.
Here we choose the distance from the origin as the collective variable.
We use PySAGES to define this collective variable.
from pysages.colvars import Distance
import pysages
# Distance from our particle to origin (particle 1)
cvs = [Distance([0, 1])]Next, we are interested in an unbiased simulation.
PySAGES offers a special method for unbiased simulations, that can record a collective variable.
from pysages.methods import Unbiased
method = Unbiased(cvs)We also want to track the collective variable over time and as a histogram, so we can log it via PySAGES.
from pysages.methods.utils import HistogramLogger
hist = HistogramLogger(period=100)We now simulate
result = pysages.run(method, generate_context, int(1e5), callback=hist)Let's see how the particle moved in this potential landscape.
def plot_cv_trajectory(result, x_range=(0, 4)):
histogram_log = result.callbacks[0]
cv_log = np.asarray(histogram_log.data)
time = np.linspace(0, 0.1 * len(cv_log), len(cv_log))
x = np.linspace(x_range[0] + 0.01, x_range[1], 200)
landscape = free_energy(energy)(x)
fig, ax = plt.subplots()
ax.set_xlabel(r"$t$ $[\tau]$")
ax.set_ylabel(r"$\xi$ $[\sigma]$")
ax.set_ylim(x_range)
ax.plot(time, cv_log, label="cv trajectory")
ax.legend(loc="center right")
ax2 = ax.twiny()
ax2.set_xlabel(r"$A(\xi)$ $[k_BT]$")
ax2.plot(landscape, x, label="energy landscape", color="orange")
ax2.legend(loc="upper left")
fig.show()plot_cv_trajectory(result)We see, that the system never leaves the local minimum around
The sampling is not ergodic! This is common for normal MD (although not as easy to spot usually).
###Solution: Biased simulations
Introducing a weight function
Find the biased distribution as a function of the collective variable
- with known distribution
$p(\xi)$ (measured) and known weight$w(\xi)$ , we can infer$p_{eq}$ - optimal weight
$w(\xi) = p_{eq}(\xi)$ : almost random walk in$\xi$ - optimal weight a priori unknown
For NVT MD we can modify the Hamiltonian to introduce a weight.
Here is where PySAGES comes into play! PySAGES allows you to easily introduce a biasing Hamiltonian into a given MD backend (like HOOMD-blue, OpenMM, or ASE). So it is not necessary to modify the MD backend and via JAX we offer automatic differentiation, so forces are calculated automatically.
We can start biasing by using a simple harmonic biasing, where we bias the system towards one of the less explored regions in phase space.
PySAGES offers a pre-defined class that implements this, which we will take advantage of.
In our example toy system, we choose
from pysages.methods import HarmonicBias
def apply_harmonic_bias(kspring, center=2, cvs=cvs, timesteps=int(1e5), log_period=100):
method = HarmonicBias(cvs, kspring=kspring, center=center)
hist = HistogramLogger(period=log_period)
result = pysages.run(method, generate_context, timesteps, callback=hist)
return resultWe don't know a priori what a good spring constant is. Let's start with
kspring = 10
result = apply_harmonic_bias(kspring)
plot_cv_trajectory(result)We observe that the free-energy barrier around
Let's try
kspring = 100
result = apply_harmonic_bias(kspring)
plot_cv_trajectory(result)Ok, now the system mostly oscillates around the local maximum, but is no longer able to come close to the actual minima of the free-energy landscape.
The spring constant is so strong, that restricts the exploration of the phase space too much. Let's try the middle ground instead
kspring = 30
result = apply_harmonic_bias(kspring)
plot_cv_trajectory(result)This looks much better!
We observe multiple transitions between the minima at
We now analyze the histograms of this trajectory to determine the free-energy landscape
from scipy import integrate
def plot_cv_histogram(result, x_range=(0, 4), bins=30):
histogram_log = result.callbacks[0]
hist, edges = histogram_log.get_histograms(bins=bins, range=[x_range])
x_hist = edges[0][:-1] + np.diff(edges[0]) / 2
weight = np.exp(-kT * kspring / 2 * (x_hist - 2)**2)
unbiased_distribution = hist / weight
unbiased_distribution /= integrate.simpson(unbiased_distribution, x=x_hist)
fig, ax = plt.subplots()
ax.set_xlabel(r"$\xi$ $[\sigma]$")
ax.set_ylabel(r"p(\xi)")
ax.set_xlim(x_range)
ax.plot(x_hist, hist, label=r"biased $p(\xi)$")
ax.plot(x_hist, unbiased_distribution, label=r"unbiased $p_{eq}(\xi)$")
ax.legend(loc="best")
fig.show()plot_cv_histogram(result)We can't be sure that this is the correct profile yet.
So let's compare to the expected free-energy profile.
def plot_free_energy(result, x_range=(0, 4), bins=30):
x = np.linspace(x_range[0] + 0.01, x_range[1], 200)
corrected_free_energy = free_energy(energy)(x)
histogram_log = result.callbacks[0]
hist, edges = histogram_log.get_histograms(bins=bins, range=[x_range])
x_hist = edges[0][:-1] + np.diff(edges[0]) / 2
weight = np.exp(-kT * kspring / 2 * (x_hist - 2)**2)
unbiased_distribution = hist / weight
unbiased_distribution /= integrate.simpson(unbiased_distribution, x=x_hist)
mask = unbiased_distribution != 0
estimated_profile = -kT * np.log(unbiased_distribution[mask])
constant_C = -np.min(estimated_profile) + np.min(corrected_free_energy)
fig, ax = plt.subplots()
ax.set_xlabel(r"$\xi$ $[\sigma]$")
ax.set_ylabel(r"A(\xi)")
ax.set_xlim(x_range)
ax.plot(x, corrected_free_energy, label=r"true $A(\xi)$")
ax.plot(x_hist[mask], estimated_profile + constant_C, label=r"estimated $A(\xi)$")
ax.legend(loc="best")
fig.show()plot_free_energy(result)That estimation is not bad. We get the approximate right shape in the middle and that could be further improved by running the sampling trajectory longer. Or try a different spring constant. [Try it out!]
But there are still some issues because we still cannot sample the entire space:
- the right and left barriers are uncovered
- the height and maximum of the sampled barrier are slightly off
- the highest local minimum is under-sampled
Can we bias simulations in these regions too, to improve sampling coverage?
We want to find the free-energy profile along a given path in the space for collective variables. Usually, this path can be multidimensional.
Example dihedral angles of Alanine Dipeptide. PySAGES Alanine Dipeptide examples
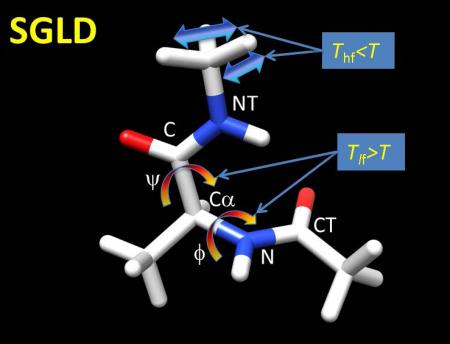
Wu, Xiongwu, Bernard R. Brooks, and Eric Vanden‐Eijnden. Journal of computational chemistry 37.6 (2016): 595-601.

Bonomi, Massimiliano, Carlo Camilloni, and Michele Vendruscolo. Scientific reports 6.1 (2016): 1-11.
Downside:
We need to know the path
Goal:
Obtain the free-energy profile along the path
Discretize the path
So we can describe the last term as a thermal average.
And the free energy is
With a single biasing potential
But if we want to combine multiple windows, we have to estimate it.
not directly solvable.
Ideal
- For now we use a harmonic potential
- fix the system with a spring to point on the path.
$$H^b_{i}(\xi) = k/2 (\xi - \xi_0(t_i))^2$$ - combine different points into one analysis
centers = np.linspace(0, 4, 10)
kspring = 100
x = np.linspace(0.01, 4, 200)
landscape = free_energy(energy)(x)
fig, ax = plt.subplots()
ax.set_xlabel(r"$\xi$")
ax.set_ylabel(r"$H_i(\xi)$")
ax.set_ylim((-12, 20))
ax.set_xlim((0, 4))
for x_c in centers:
label = "biasing potential" if x_c == 4 else None
ax.plot(x, kspring / 2 * (x - x_c)**2, label=label)
ax.plot(x, landscape, label="potential")
ax.legend(loc="best")
fig.show()- one free parameter per window
$k$ Goal:$\Rightarrow$ overlapping histograms
from pysages.methods import UmbrellaIntegration
method = UmbrellaIntegration(cvs, kspring, centers, 100)
result = pysages.run(method, generate_context, int(1e5))You can already see one downside, we have to run multiple replicas, which consume more computational resources.
Let's see what the histograms look like.
def plot_multi_histogram(result, x_range=(0, 4), bins=30):
xs = []
histograms = []
for histogram_log in result.callbacks:
hist, edges = histogram_log.get_histograms(bins=bins, range=[x_range])
xs.append(edges[0][:-1] + np.diff(edges[0]) / 2)
histograms.append(hist)
fig, ax = plt.subplots()
ax.set_xlabel(r"$\xi$ $[\sigma]$")
ax.set_ylabel(r"p(\xi)")
ax.set_xlim(x_range)
for x, hist in zip(xs, histograms):
ax.plot(x, hist, label=r"biased $p(\xi)$")
fig.show()plot_multi_histogram(result)The result is OK, good enough for our test case here. We have some overlap between the histograms.
Now, how can we combine these histograms into a single free-energy profile?
One option is weighted histogram analysis (WHAM), but that works best for one-dimensional collective variables. (Implement it yourself, all you need from PySAGES is available.)
Here we are using a simple umbrella integration instead.
Idea: differentiate by
For this we approximate
approximating up to second order gives us
Now we can combine all these windows
And integrate the derivative for the profile numerically.
- this averages the mean-force
- we are approximating the distribution
- easy to generalize in higher dimensions
Let's see how PySAGES analyzes it for us and produces the free-energy result.
def plot_umbrella_free_energy(pre_result, x_range=(0, 4)):
x = np.linspace(x_range[0] + 0.01, x_range[1], 50)
landscape = free_energy(energy)(x)
result = pysages.analyze(pre_result)
centers = np.asarray(result["centers"])[:, 0]
estimate = np.asarray(result["free_energy"])
estimate = estimate - np.min(estimate) + np.min(landscape)
fig, ax = plt.subplots()
ax.set_xlabel(r"$\xi$ $[\sigma]$")
ax.set_ylabel(r"A(\xi)")
ax.set_xlim(x_range)
ax.plot(x, landscape, label=r"true $A(\xi)$")
ax.plot(centers, estimate, label=r"estimated $A(\xi)$")
ax.legend(loc="best")
fig.show()plot_umbrella_free_energy(result)Even with the crude finite-differences approximations we are doing we can estimate the shape of the potential.
Just the second minimum is underestimated, which could be fixed with more sampling and more sampling points in that vicinity. [Try it out!]
Difficulties:
- choose a good spring constant
- if it is too large, the histograms won't overlap
- if it too small, you won't be able to sample some barriers
- choose a good number of replicas
Can we do better than this? Yes, of course:
- Meta-dynamics: approximate one weight function with a sum of Gaussians
- ANN: approximate biasing force with artificial neuronal networks (ANN)
- And many more sampling methods implemented in PySAGES
This example was using a single CPU core only. For a realistic system, GPU acceleration is required.
What do we have to do to run this code on a GPU?
Nothing!
Just change the runtime environment to GPU and you are good to go. Or run your examples on the GPU accelerated workstation.
PySAGES is preinstalled, so you are good to go!
We can determine the profile of free energy now. In more than one dimension the profile depends on the path taken.
Bonomi, Massimiliano, Carlo Camilloni, and Michele Vendruscolo. Scientific reports 6.1 (2016): 1-11.
So how do we find the minimum (free) energy path (MEP)?
This path runs through a valley of the free-energy landscape and has the smallest free-energy barrier. Hence it is the most likely kinetic path.
The formal definition of this path is that the forces on a state point are always parallel (up or down not sideways) to the path.
General Idea:
Move the string (path) along the perpendicular component
Problem 1:
How to get
Solution: Harmonic biased simulations at each replica of the string
[computationally expensive]
Problem 2:
How to get the perpendicular component?
- the discretization of the path is rough
Solution: use a cubic spline-interpolation
- piece-wise 3rd order polynomial
- easy to parameterize
- easy to interpolate
- differentiable
Using the perpendicular component directly does not lead to good convergence. Instead, we can use the improved string method.
Iterate:
- Calculate
$\Delta V|_{t_i}$ - Move string
$\xi_0(t_i)$ +=$\alpha \Delta V|_{t_i}$ - Calculate spline
$\xi_0^s(t)$ - Re-parametrize the string points such that
$|\xi_0^s(t_i) - \xi_0^s(t_{i+1})| = const$ .- cancels out parallel force component
- well-discretized string
- requires a norm in
$\xi$ space. For example$L^2$ norm in$\xi$ space.
Stop iteration if converged and test
- typically slow convergence
With the converged path, calculate the free-energy profile using umbrella integration.
Let us define a committor probability.
For a given point
This is a probability since we can have multiple realizations of
Intuitively, if
fig, ax =plt.subplots()
ax.set_xlabel("$t$")
ax.set_xlim((0,1))
ax.set_ylabel("$ p_B(t)$")
x = np.linspace(0, 1, 50)
p_good = np.tan(2*(x-.5))
p_good -= np.min(p_good)
p_good /= integrate.simpson(p_good, x=x)
p_bad = x*0+1
p_bad /= integrate.simpson(p_bad, x=x)
ax.plot(x, p_good, label="good path")
ax.plot(x, p_bad, label="bad path")
ax.legend(loc="best")
fig.show()We can generalize this concept to the entire space.
First, the isosurfaces of the collective variable are all points in space
We can also define isosurfaces for the committor probabilities.
All points in space where the committor probabilities are equal
For a descriptive collective variable, we want that when changing
Usually, calculating committor probabilities is computationally so expensive that it is prohibitive. But it is a way to check results in case of doubt about collective variables.