You signed in with another tab or window. Reload to refresh your session.You signed out in another tab or window. Reload to refresh your session.You switched accounts on another tab or window. Reload to refresh your session.Dismiss alert
File endings .fastq | .fq | .fastq.gz | .fq.gz are now removed from the output file (unless they were specified with --basename) in a bid to reduce the length of the already long file names.
Enabled the new option --dovetail (which will be turned on by default for --pbat libraries) which will now allow dovetailing reads to be reported. For a more in-depth description see #14.
Changed the behaviour of corner cases to where several non-directional alignments could have existed for the very same position but to different strands so that now the best alignment trumps the weaker one. As an example: If you relaxed the alignment criteria of a given alignment to allow ~60 mismatches for PE alignment we did find an alignment to the OT strand with a combined AS of -324, but there also was an alignment to the CTOB strand with and AS of 0 (perfect alignment). The CTOB now trumps the OT alignment, and the methylation information information is now reported for the bottom strand. Credits go to Sylvain Foret (ANU, Canberra) for bringing this to our attention!
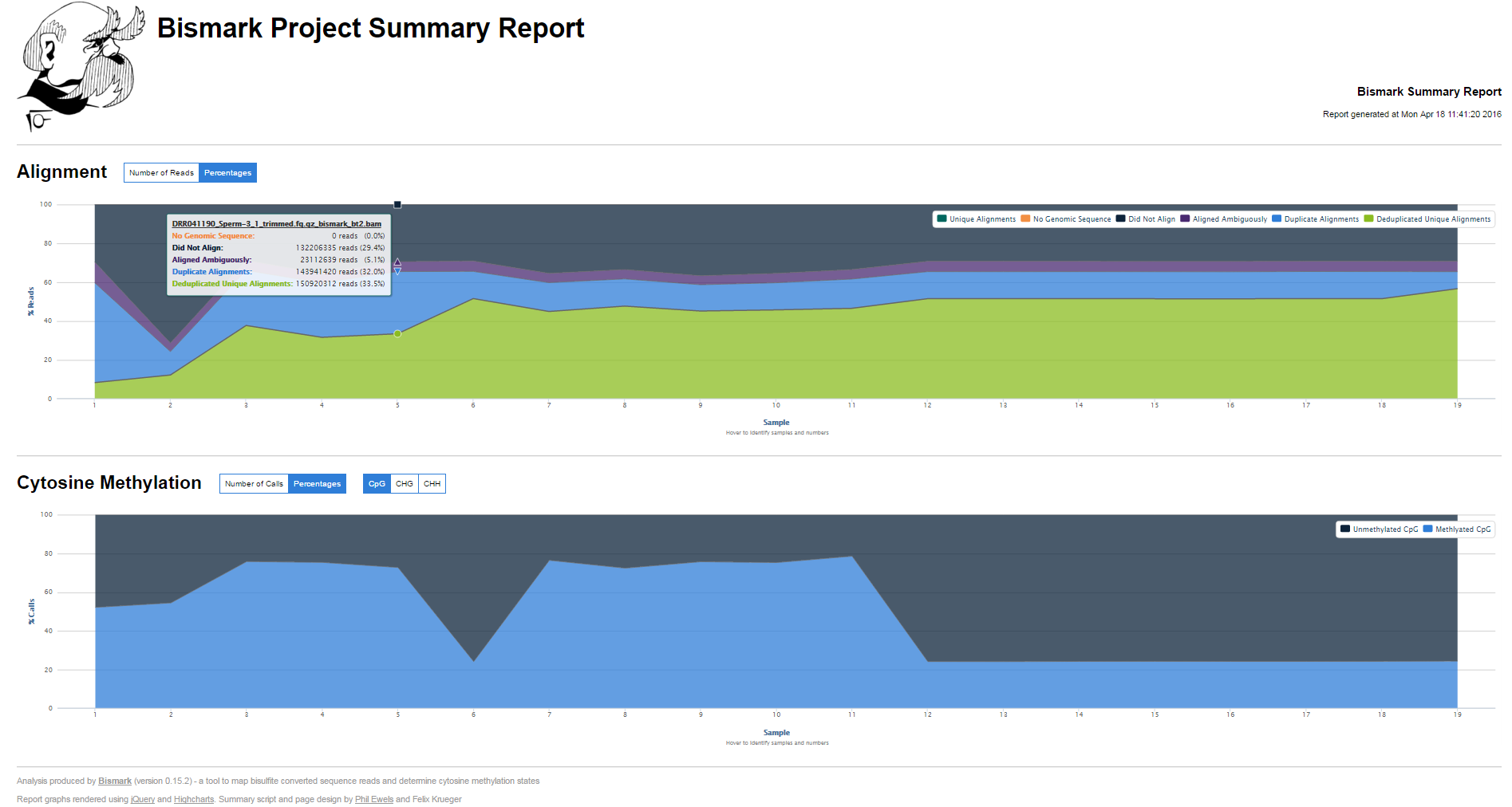
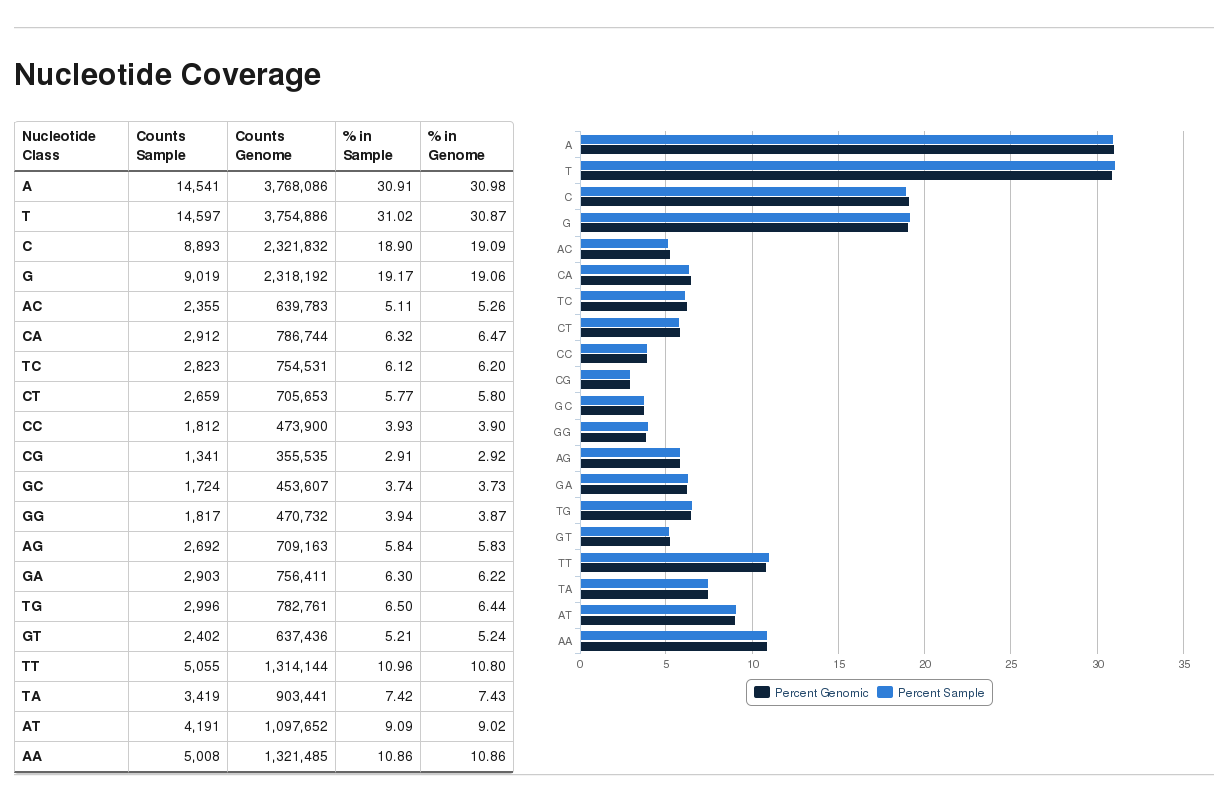
The new Bismark module bam2nuc calculcates the average mono- and di-nucleotide coverage of libraries and compares this to the genomic average composition. bam2nuc can be called straight from within Bismark (option --nucleotide_coverage) or run stand-alone. bam2nuc creates a ...nucleotide_stats.txt file that is also automatically detected by bismark2report and incorporated into the HTML report.
bismark2_sitrep.tpl
Removed an extra function call in bismark_sitrep.tpl so that the M-bias 2 plot is drawn once the M-bias 1 plot has finished drawing (parallel processing could with certain browsers and data may have resulted in a white spaceholder only).
methylation extractor
Altering the file path handling of coverage2cytosine and bismark2bedGraph also required some changes in the methylation extractor.
bismark2bedGraph
Input file path handling has been completely reworked. The output file which can be specified as -o output.bedGraph now has to be a single file name and mustn't contain any path information. A particular output folder may be specified with -dir /any/path/.
Addressing the file path handling issue also fixed a similar issue with the option --remove_spaces when -o had been specified.
coverage2cytosine
Changed zcat for gunzip -c when reading a gzipped coverage file. This should avoid some Mac platforms crashing because zcat invariably requires a file to end in the .Z (which it doesn't...)
Changed the way in which the coverage input file is handed over from the methylation_extractor
to coverage2cytosine (previously the path information might have been part of the file name, but
instead it will now be only part of the -dir output_directory option.