Phasing for metagenomics using PacBio long reads
Current Version (07/13/2021): MagPhase v1.0
MagPhase is for phasing of metagenomics data using long reads.
MagPhase is a modified version of IsoPhase which was originally designed for isoform-level phasing of PacBio Iso-Seq (full-length transcript sequencing) data.
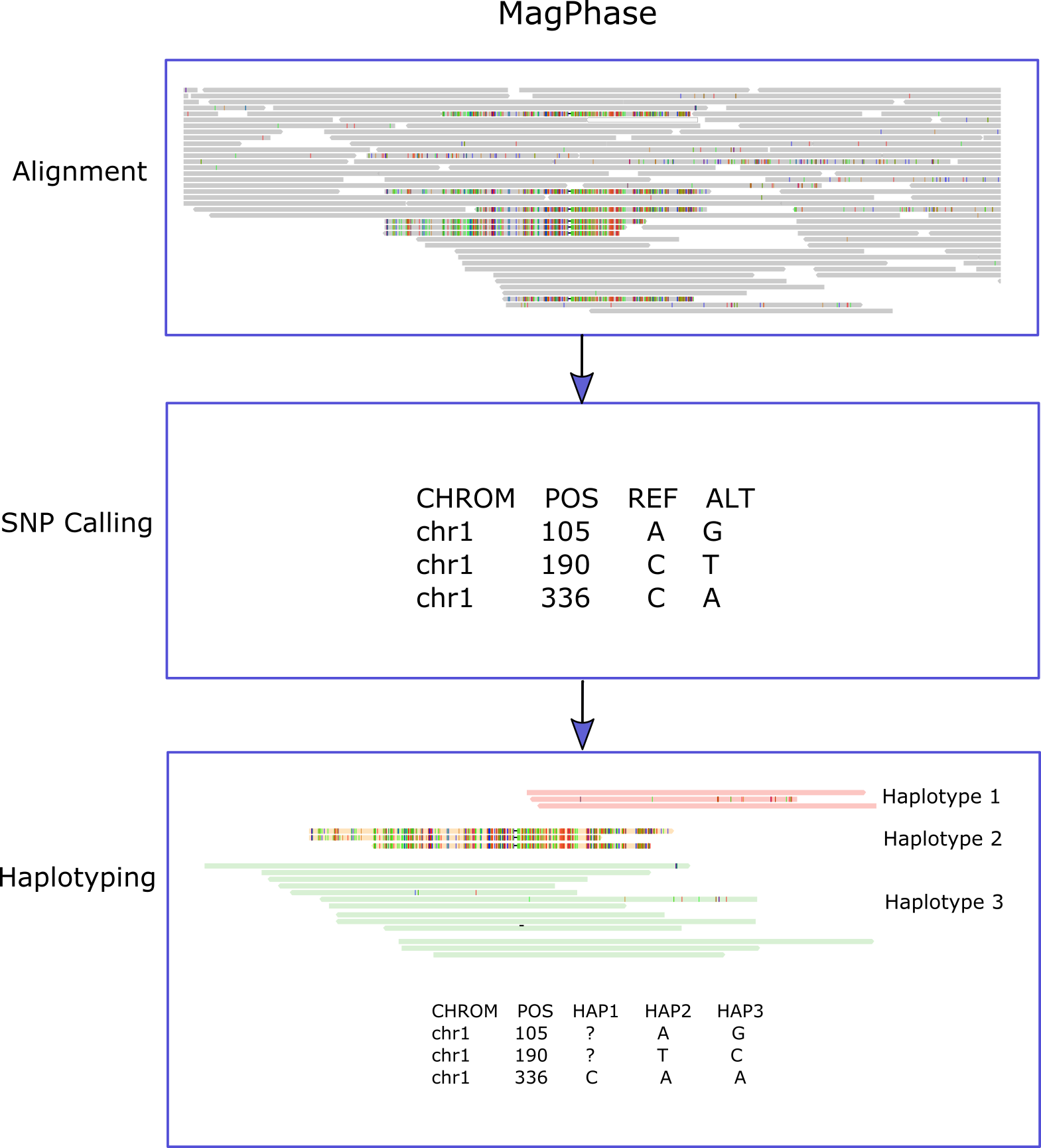
MagPhase takes an alignment BAM file of HiFi reads aligned to the assembled contigs and a BED file that denotes the regions to phase.
For each region, individual SNPs are called. Then, reads are used to infer the "haplotypes" (or lineages).
The output of MagPhase consists of individual SNP information and the inferred list of haplotypes.
- Python (3.7+)
- minimap2
- biopython
- bx-python
- scipy
- pysam
- pyvcf
We recommend using Anaconda to set up your conda environment. Currently only Linux environments are supported.
(1) Install Conda Environment
export PATH=$PATH:<path_to_anaconda>/bin
conda -V
conda update conda
(2) Clone the Github repo and install using the yml script
git clone https://github.com/Magdoll/MagPhase.git
cd MagPhase
conda env create -f MagPhase.conda_env.yml
source activate MagPhase.env
(3) Once you have activated the virtual environment, you should see your prompt changing to something like this:
(MagPhase.env)$
(4) Compile and install MagPhase
(MagPhase.env)$ python setup.py build
(MagPhase.env)$ python setup.py install
The usage for mag_phaser.py is as follows:
$ mag_phaser.py -h
usage: mag_phaser.py [-h] -a ASSEMBLY -b BAMFILE -o OUTPUT -g GENES [-p PVAL_CUTOFF] [--bhFDR BHFDR]
optional arguments:
-h, --help show this help message and exit
-a ASSEMBLY, --assembly ASSEMBLY
The mag assembly file in fasta format
-b BAMFILE, --bamfile BAMFILE
Aligned reads in bam file format [full path needed!]
-o OUTPUT, --output OUTPUT
output prefix
-g GENES, --genes GENES
SCG gene bed file
-p PVAL_CUTOFF, --pval_cutoff PVAL_CUTOFF
P value cutoff for variant calls
--bhFDR BHFDR FDR to be used for the Benjamini–Hochberg correction. Default: None (not used).
where -a provides MAG assembly contig fasta file. -b provides the aligned HiFi reads to the contig fasta. -g provides a BED file that contains the individual regions to be phased.
-p and --bhFDR controls the p-value cutoff for SNP calling. It is recommended that you use the Benjamini–Hochberg correction for better SNP detection (since correction for multiple testing can drastically reduce number of SNPs called). We recommend using --bhFDR 0.01 for general metagenomics applications.
An example run:
mag_phaser.py -a all_contigs.fasta -b all_contigs.bubbles.ccs.filtered.sorted.bam -g 1377.shortmaps.bed --bhFDR 0.01 -o 1377.strain
will produce the following files:
1377.strain.human_readable_by_hap.txt
1377.strain.human_readable_by_pos.txt
1377.strain.human_readable_by_read.txt
Within a region (as provided by the -g BED file), if there were phasing results (note: note all regions can be phased, as there could be no SNPs present), the haplotypes are represetned as a string of concatenated SNPs. For example, if there are three SNPs in this region at genomic position 101, 150, and 220, and there are four haplotypes, then the representation in the _hap.txt would be:
| haplotype | hapIdx | contig | count |
|---|---|---|---|
| ATT | 0 | contig_1337 | 10 |
| CTG | 1 | contig_1337 | 12 |
| ATG | 2 | contig_1337 | 2 |
| ?AG | 3 | contig_1337 | 4 |
Note not all haplotypes will cover all SNP positions, so some haplotypes may have a ? indicating lack of bases at that SNP location.
The SNP position would be stored in the _pos.txt file:
| haplotype | contig | pos | varIdx | base |
|---|---|---|---|---|
| ATT | contig_1337 | 101 | 1 | REF |
| ATT | contig_1337 | 150 | 2 | REF |
| ATT | contig_1337 | 220 | 3 | REF |
| CTG | contig_1337 | 101 | 1 | ALT0 |
| CTG | contig_1337 | 150 | 2 | REF |
| CTG | contig_1337 | 220 | 3 | ALT0 |
| ATG | contig_1337 | 101 | 1 | REF |
| ATG | contig_1337 | 150 | 2 | REF |
| ATG | contig_1337 | 220 | 3 | ALT0 |
| ?AG | contig_1337 | 150 | 2 | ALT0 |
| ?AG | contig_1337 | 220 | 3 | ALT0 |