The YAMP workflow
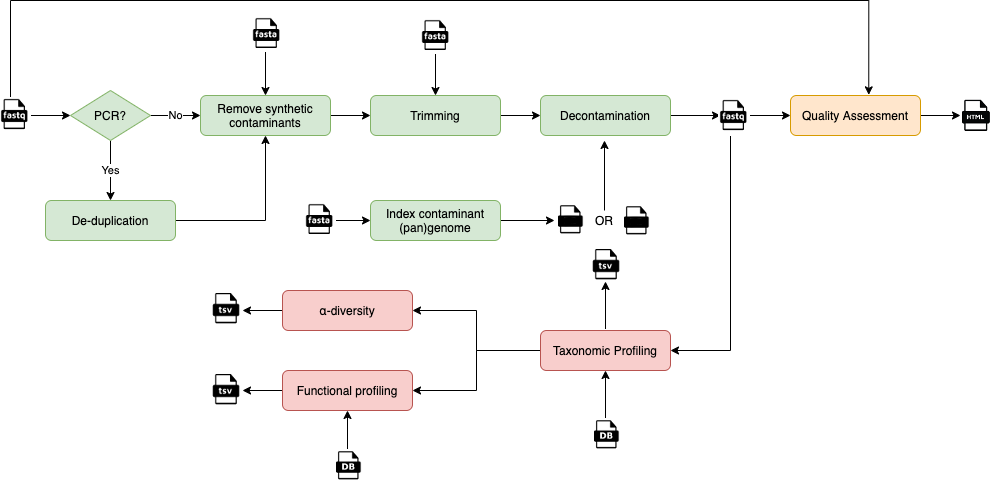
The image above depicts the key steps in the analysis of a metagenomic sample.
More in details, the QC (green blocks, performed using several tools from the BBmap suite) allows de-duplication, trimming, and decontamination of metagenomics sequences. Visualisation of the data quality is provided for the raw and QC'd reads (orange block, performed using FastQC).
The QC is followed by multiple steps aiming at characterising the taxonomic and functional diversity of the microbial community (pink blocks). Taxonomic binning and profiling is performed through MetaPhlAn, which uses clade-specific markers to both detect the organisms present in a microbiome sample and to estimate their relative abundance. The functional capabilities of the microbiome community are currently assessed by the HUMAnN pipeline which first stratifies the community in known and unclassified organisms using the MetaPhlAn's results and the ChocoPhlAn pan-genome database, and then combines these results with those obtained through an organism-agnostic search on the UniRef proteomic database. QIIME2 is used to evaluate multiple diversity measures.
YAMP can be run in three modes:
-
complete, from the raw data up to the functional annotation (green, orange, and pink blocks), -
QC: limiting the analysis to the QC steps (green and orange blocks), or -
characterisation: limiting the analysis to the characterisation of the taxonomic and functional diversity (pink blocks).