{kind=link}
Supporting repository for article "Software Evaluation for 'de novo' Detection of Transposons"
Datasets used for testing the TE de novo detection tools:
| Dataset | Sequences | Length |
|---|---|---|
| Human genome | chromosome 21 | 46.7 Mb |
| Zebrafish | chromosome 1 | 137.5 Mb |
| Fruit fly | whole genome | 137.6 Mb |
| Simulated data | simulation | 100.1 Mb |
Requirements:
- Python 3.x
- Biopython
- NumPy
- PyYAML
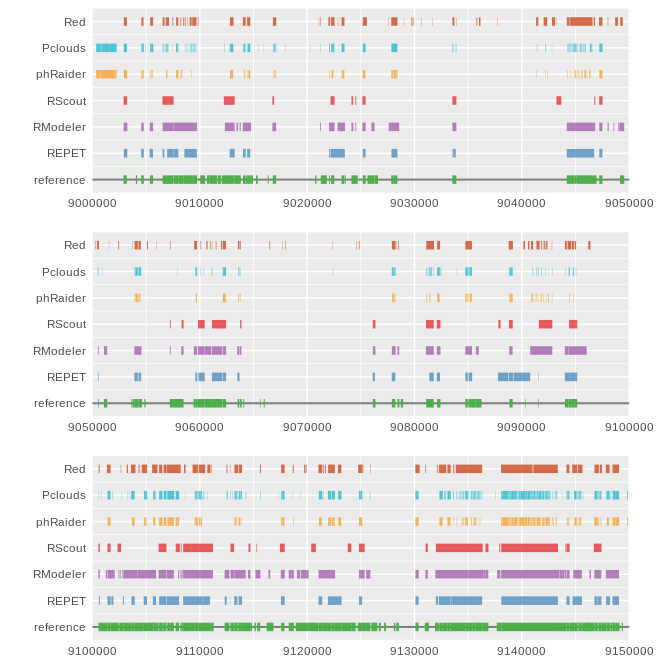
repeat_analysis.py : script that compares the gff of an annotation to a reference and outputs the percent annotated for each entry, a global confusion matrix and the Matthews Correlation Coefficient
e.g.: ./repeat_analysis.py reference.gff repeats.gff
random_sequence_TEs.py : script needs config.yml and the files specified there (repeats_list and repeats.fa). Outputs a fasta sequence with random TEs and an annotation file (GFF).
random_nest_TEs.py : script run after random_sequence_TEs.py, requires the same files. Outputs a fasta sequences with nested TEs and an updated GFF file.
repeats_list : configuration file with a row for each TE and 9 columns: id_of_TE, number_of_repeats, %_identity, std_dev, %_indels, has_TSD?, real_length, %_fragmentation, %_nested
config.yml : configuration file with prefixes for the output, size of base sequence, location of repeats_list and fasta files, gc_content and seed (optional).
First run inserts TEs in base sequence:
e.g.: ./random_sequence_TEs.py (expects repeats.fa, config.yml and repeats_list in same directory)
output: prefix_out_repeats.gff, prefix_out_repeats.fasta (only modified repeats), prefix_out_sequence.fasta (full sequence)
Next run nests TEs in the previous sequence:
e.g.: ./random_nest_TEs.py (expects repeats.fa, config.yml, repeats_list, prefix_out_repeats.gff in same directory)
output: prefix_out_repeats_nest.gff, prefix_out_sequence_nest.fasta (full sequence)
Requirements:
- R
- ggplot2
- gridExtra
gff_tracks.R: can be manually edited to change the path of the GFF files to be plotted.