
v4.5 - Somehow Isoform Returned
Takes R2C2/C3POa and/or PacBio/ccs/lima data and defines high confidence isoform consensus sequences and alignments. You can mix and match R2C2/PacBio reads and fasta/fastq files (quality scores are ignored). Mandalorion is not tested for use with regular ONT reads. I recommend using FLAIR instead for those.
to trim polyA tails and or adapter sequences:
Look at the reads in your hypothetical input.5to3.fasta file.
If your reads look like this (some untrimmed adapter sequences, polyA tail still present)
CATGTGTATAAGAGACAGTAGTACTTGGTGGGG [RNA transcript 1] AAAAAAAAAAAAAAAAAAAAAAGTGAACTGAGCGACTCTGCGT
CATGTGTATAAGAGACAGTAGTACTTGGTGGGG [RNA transcript 2] AAAAAAAAAAAAAAAAAAAAAAGTGAACTGAGCGACTCTGCGT
CATGTGTATAAGAGACAGTAGTACTTGGTGGGG [RNA transcript 3] AAAAAAAAAAAAAAAAAAAAAAGTGAACTGAGCGACTCTGCGT
|-------------33nt--------------| |--------21nt-------|
remove untrimmed adapter sequences and polyA tails like this:
python3 Mandalorion/utils/removePolyA.py -i input.5to3.fasta -o input.5to3.noPolyA.fasta -t 33,21
after trimming adapters and polyA sequences:
python3 Mandalorion/Mando.py -p ./ -g gencodeV29.gtf -G hg38.fasta -f input.5to3.noPolyA.fastawill generate
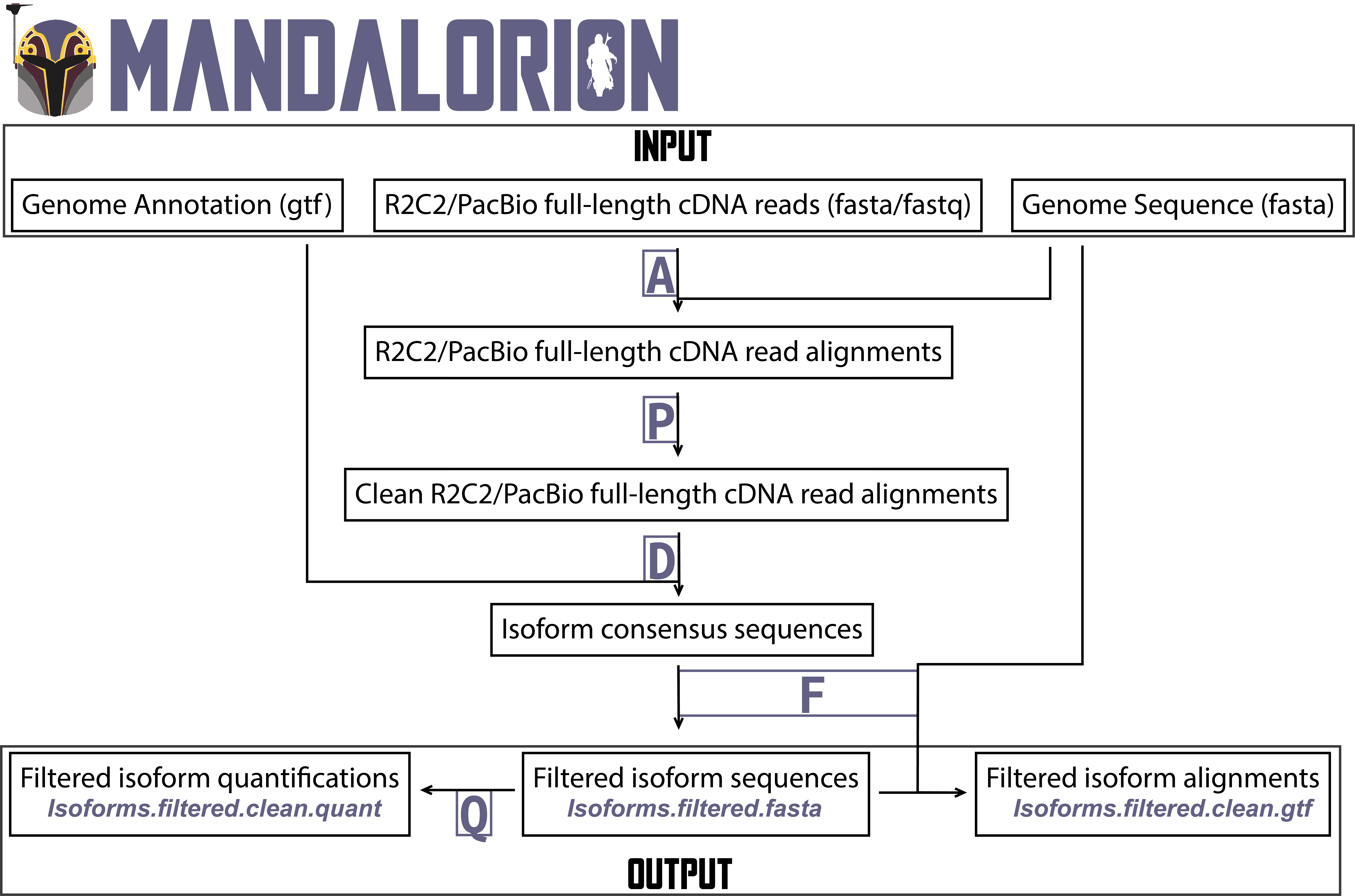
Isoform sequences (.fasta), models (.gtf,.psl), and quantification, as well as isoform feature (TSS, PolyA, splice junction) quantification (.quant,.rpm)
Mandalorion contains several modules that are run sequentially by default (APDFQ) to define, align, and quantify high-quality isoforms.
Just navigate to the folder you want Mandalorion to live and git clone the repository
git clone https://github.com/christopher-vollmers/Mandalorion.gitMake sure a C++ compiler installed, then run the following to install all dependencies for Mandalorion
cd Mandalorion
chmod +x setup.sh
sh setup.shThe command installs local copies of
as well as use pip install to install the correct versions of
This version was tested with v1.4.1 of abpoa.
It is best to run Mandalorion with reads that have no adapter sequences (trimmed), no polyA tails, and are in 5'->3' orientation. By default, C3POa (v3) does trim reads but doesn't do polyA removal and, depending on the adapters used, may not put reads in 5'->3' orientation..
Therefore the removePolyA.py script in utils/ should be used to get both PacBio (depending on preprocessing) and R2C2 reads ready.
The script first removes a fixed number of bases (can't be zero) from the ends of the read and then removes the polyA tail on the 3' end of the read. Depending on the polyA position it will also reorient the read in 5'->3' direction.
You can call Mando.py from the Mandalorion folder or from anywhere else so either
cd /path/to/Mandalorion/
python3 Mando.py [OPTIONS]or
python3 /path/to/Mandalorion/Mando.py [OPTIONS]Running with default settings:
python3 Mando.py -p . -g gencodeV29.gtf -G hg38.fasta -f Consensus_reads.fofn.fofn file structure is simply a text file with one line per input fasta/fastq file. You can mix and match fasta/fastq files and gzipped and unzipped files.
/path/to/file1.fasta
/path/to/file2.fastq
/path/to/file4.fastq.gzHere is a full list of options that you can use to modify Mandalorion behavior:
-h, --help show this help message and exit
-p PATH, --path PATH Directory to put output files into
-u UPSTREAM_BUFFER, --upstream_buffer UPSTREAM_BUFFER
Defines upstream leniency window for polyA and TSS
determination (default 10)
-d DOWNSTREAM_BUFFER, --downstream_buffer DOWNSTREAM_BUFFER
Defines downstream leniency window for polyA and TSS
determination (default 50)
-g GENOME_ANNOTATION, --genome_annotation GENOME_ANNOTATION
Genome annotation file (gtf). Is used to identify
individual annotated splice sites. If -W is set it is
also used to white-list annotated polyA sites
-G GENOME_SEQUENCE, --genome_sequence GENOME_SEQUENCE
Genome file (fasta)
-r MINIMUM_RATIO, --minimum_ratio MINIMUM_RATIO
Proportion of reads that align to a locus required for
an isoform (default 0.01)
-i MINIMUM_INTERNAL_RATIO, --minimum_internal_ratio MINIMUM_INTERNAL_RATIO
-R MINIMUM_READS, --minimum_reads MINIMUM_READS
Minimum number of reads to make an isoform (default 3)
-f CONSENSUS_READS, --Consensus_reads CONSENSUS_READS
Fasta/fastq file with R2C2/PacBio consensus reads, can
be entered as a single file path, a comma separated
list of file paths, or a path to a file of filenames
file (has to end on .fofn) that contains one file path
per line
-O OVERHANGS, --overhangs OVERHANGS
Defines bounds for unaligned bases on ends. Format:
min5prime,max5prime,min3prime,max3prime (default
0,40,0,40)
-t MINIMAP2_THREADS, --minimap2_threads MINIMAP2_THREADS
Number of threads to use when running minimap and
consensus calling (default 4)
-I MINIMUM_ISOFORM_LENGTH, --minimum_isoform_length MINIMUM_ISOFORM_LENGTH
Minimum length in nt of isoforms that will be
considered (default 200)
-n MINIMUM_FEATURE_COUNT, --minimum_feature_count MINIMUM_FEATURE_COUNT
Features (starts,ends, new splice sites) will be
considered if they are present in this number of reads
(default 2)
-w SPLICE_SITE_WINDOW, --splice_site_window SPLICE_SITE_WINDOW
reads spliced within this number of nucleotides on
each side of a splice site will be considered spliced
at this site (default 1)
-A ACUTOFF, --Acutoff ACUTOFF
Isoforms with A content of more than this cutoff in a
30nt window surrounding their polyA site will be
discarded (default 0.5)
-W WHITE_LIST_POLYA, --white_list_polyA WHITE_LIST_POLYA
If set, polyA sites that fall within +/-20nt of
annotated transcript ends will not be filtered
regardless of Acutoff set with -A. Annotated
transcript ends will be taken from annotation file
given with -g. Only transcripts having one of the
provided comma separated values in the line of their
exon features will be used. E.g. setting -W to
[SIRV,"basic"] will whitelist spike-in SIRV
transcripts and transcripts considered "basic", i.e.
high confidence full length, in the gencode
annotation. Setting -W to [exon] should include all
transcripts in the gtf file. This is feature only
checks whether the provided values are in the line,
not where they are. That means that setting -W to
[chr1] will whitelist all transcripts on chromosome 1
but also transcripts with "chr1" in their name, so
it's a bit dangerous
-m, --multi_exon_only
If used, Mandalorion will filter all single exon
isoforms
-j JUNCTIONS, --junctions JUNCTIONS
base context required (in lower-case for either
strand) to call unannotated splice junctions (default
gtag,gcag,atac,ctac,ctgc,gtat)
-M MODULES, --Modules MODULES
Defines what modules of Mandalorion will be run. By
default this includes: A - Alignment, P - .sam to
.clean.psl conversion, D - defining isoforms, F -
Filtering isoforms, Q - Quantifying isoforms. Each
module needs the output of the modules run before it
to function properly. Running individual modules can
be useful if you want to for example filter with
different parameters without rerunning the whole
pipeline. (default APDFQ)
-v, --version Prints Mandalorion version
Isoforms.filtered.fasta: The consensus sequence of each high-confidence isoform.
Isoforms.filtered.clean.gtf: Alignments of the isoform consensus sequences with small indels removed
Isoforms.filtered.clean.quant: Number of reads from each supplied fasta file associated with each isoform
Isoforms.filtered.clean.rpm: Normalized expression values for each supplied fasta file associated with each isoform
Isoforms.filtered.clean.genes: Isoform groups and gene association
TSSs.quant: Number of reads from each supplied fasta file associated with each TSS
TSSs.rpm: Normalized expression values for each supplied fasta file associated with each TSS
PolyAs.quant: Number of reads from each supplied fasta file associated with each PolyA site
PolyAs.rpm: Normalized expression values for each supplied fasta file associated with each PolyA site
Junctions.quant: Number of reads from each supplied fasta file associated with each splice junction
Junctions.rpm: Normalized expression values for each supplied fasta file associated with each splice junction
These are the scripts used to do haplotype phasing and HLA analysis as well as preparing reads for Mandalorion and parsing output to fit the LRGASP consortium requirements. There is also a script to plot a genome browser shot as shown here.
