{kind=link}
This repository demonstrates the modular approach we propose on how to organize and develop Nextflow workflows via shareable and reusable packages using the WFPM CLI tool (https://github.com/icgc-argo/wfpm).
This is a simplified version of the ARGO DNA Alignment Workflow (https://github.com/icgc-argo/dna-seq-processing-wfs). However the underlying
tools bwaMemAligner, bamMergeSortMarkdup and cleanupWorkdir are mostly the same (much of the
code was simply copied over here) as the ARGO productoin workflow.
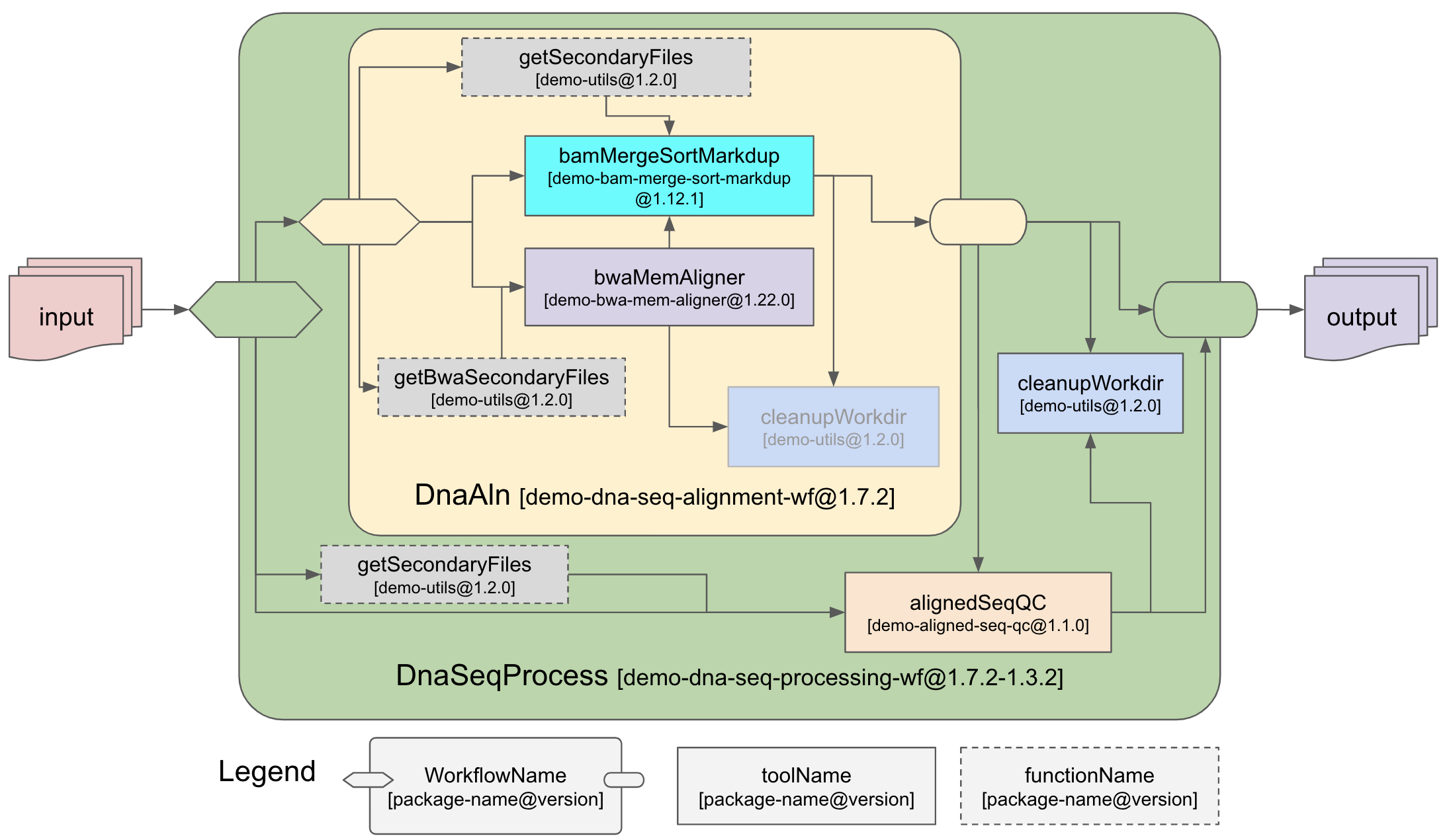
As long as you have input sequencing data (lane level unmapped BAMs) and reference genome files, and able to run Docker, you can run the workflow locally. For testing purposes, this repository contains small sequencing and reference genome files. The easiest way to try out the workflow is to clone the repo and launch test runs using commands as below:
git clone https://github.com/icgc-argo/demo-wfpkgs.git
cd demo-wfpkgs
nextflow run icgc-argo/demo-wfpkgs/demo-dna-seq-processing-wf/main.nf -r demo-dna-seq-processing-wf.v1.7.2-1.3.2 -params-file test-1.nf.json
The output of aligned lane BAMs, merge / markduplicated CRAM and QC metrics will be under the
outdir folder. The content should be something like:
outdir/
├── DnaSeqProcess_DnaAln_bwaMem
│ ├── grch38-aligned.AAA.227005013f41989b8719d3dfcdf1e105.lane.bam
│ ├── grch38-aligned.AAA.b98a338e747f0a01d23b699bdc103dab.lane.bam
│ └── grch38-aligned.BBB.09149666597ac5f96e53990f4d23ef04.lane.bam
├── DnaSeqProcess_DnaAln_merSorMkdup
│ ├── grch38-aligned.merged.cram
│ ├── grch38-aligned.merged.cram.crai
│ └── grch38-aligned.merged.duplicates_metrics.tgz
└── DnaSeqProcess_alignedSeqQC
├── grch38-aligned.merged.cram.bamstat
├── grch38-aligned.merged.cram.extra_info.json
├── grch38-aligned.merged.cram.qc_metrics.tgz
├── grch38-aligned.merged.cram_AAA.bamstat
├── grch38-aligned.merged.cram_BBB.bamstat
└── grch38-aligned.merged.cram_C0HVY-AAA.bamstat
More test params files are available under demo-dna-seq-processing-wf/tests with name
pattern: test-*.nf.json.