haplotype.r is a command-line R script for generating haplotype network plots with minimum fuss. It was written on a Mac and as-such probably only works on a Mac, but you're welcome to try it on other platforms. I wrote this because I often found there was a maximum of fuss when doing this and I wanted a more general-case, easy-to-use thing. It works great for me, I hope it works great for you.
haplonet.r relies upon the following dependencies:
-
littler (specifically, you need an executable called littler in your path for this to work. On my system, the homebrew version of littler did the trick, but if you install it some other way, just note that you'll have to rename the executable, make a link to the original that's called littler, or change the hashbang line in the script itself.)
-
pegas, or alternatively, use my modified version of pegas to overcome the issue (mentioned at the bottom of this document) where mutation tick marks are incorrectly displayed on haplotype links.
-
beyonce (This last is required as-written for color palette generation, but the script can easily be re-tooled for other color palettes. For more R color packages, see here)
Currently, haplonet.r is written using the quartz graphics device, which limits it to Mac systems, but subsequent versions may support other devices.
At minimum, haplonet.r requires you to have a sequence alignment in FASTA format and a text-based, delimited data file (the default delimiter is tab, but can be set via command-line options. The data file must have a field called 'id' that matches the FASTA sequence identifiers and a field that defines some category which by default is named 'region' but can be changed in the options.
Example alignment:
>seq1
GATTACA
>seq2
ATTACA-
>seq3
TACCAGA
Example data file:
| id | marker | region | country |
|---|---|---|---|
| seq1 | coi | northwest | Kiribati |
| seq2 | coi | southeast | Kiribati |
| seq3 | coi | east | Kiribati |
The order of sequences in either the data or alignment files is not important as the script associates samples by sequence identifier.
So, with a sequence alignment of one species and a data file describing (say) where they were sampled, you'd do:
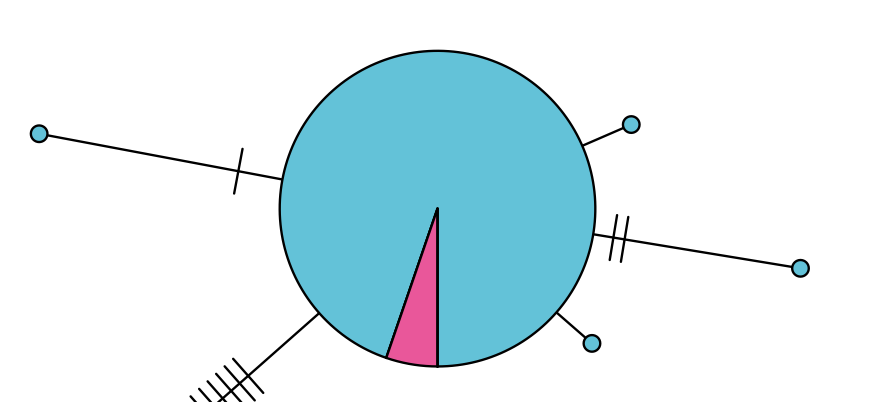
$ ./haplonet.r alignment.fasta data.tabIf everything worked properly, you should get a graphics window popping up with a likely shoddy-looking haplotype network, like so:

This window has popped up in interactive mode, allowing you to rearrange the various haplotypes until the plot looks good. To do this, simply click on a haplotype, then click where you want it to end up. Once you're done doing this, right-click somewhere on the plot and it will close and save to a PDF called by default hapnet_plot.pdf.
After a bit of tweaking, the new plot may look something like this:

Usage:
haplonet.r [--separator=<sep>] [--filter=<filter>] [--order-categories=<cats>] [--field=<field>] [--output=<file>] [(--legend --legend-position=<pos> [--save-legend])] [--haplotype-labels] [--big-palette] [--dump] <alignment> <datafile>
Options:
-s --separator=<sep> Data file field separator [default: tab]
-f --field=<field> Category field in data file [default: region]
-t --filter=<filter> Filter taxa by pattern
-c --order-categories=<cats> Specify category order (fmt: x,x,x,x)
-o --output=<file> Output file prefix [default: hapnet]
-l --legend Print a legend
-p --legend-position=<pos> Legend position [default: topleft]
-v --save-legend Optional second file to save legend by itself
-h --haplotype-labels Display haplotype labels
-b --big-palette Use large color palette for many categories
-d --dump Dump the options for debuggingAllows you to specify how your data file is delimited. This defaults to tab but you can pass any character or string that is supported by the read.table function in R.
option:
-s --separator=<sep>
Determines which field in your data file is used when drawing colored pies in the haplotype plot. The default value for this is a field called 'region'. May be specified as an index (1-based) or a field name.
option:
-f --field=<field>
This option allows you to subset sequences by a search pattern. For example, if your alignment/data file contains more than one species and you want to generate a network comprising a single species, this is the option for you. There are two ways to do this. If you pass a search pattern (regular expressions are supported as-is), the script will filter the sequences by sequence identifier. If you pass a string or integer, followed by a colon, followed by a search patttern (e.g. "species:vanderbilti"), it will treat the word or number before the colon as the data column it should filter and the pattern after the color as the search criteria. Thus, passing --filter=Cvd would search sequence ID's for the string 'Cvd', whereas --filter=species:vanderbilti would search the column called species in the data file for the string 'vanderbilti'. The bit before the colon may also be a number, which specifies the (1-based) index of the data column you want to filter.
option:
-t --filter=<filter>
Different levels of data categories are by default shown in alphabetical order. To rearrange them, use this option. Pass a comma-separated list of your category levels in the order you would like them to appear. For this to work, you must pass all of the category levels.
option:
-c --order-categories=<cats> (fmt: x,x,x,x)
Set the prefix for output filenames. The default is hapnet, but you can set it to whatever.
option:
-o --output=<file>
This option allows you to create a legend for your haplotype network. Right now, there's not a lot of customization available, but you can always customize to your heart's content using the R data objects saved by the script. There are a few options that may or may not be required at the same time:
options:
-l --legend Specify that you want a legend
-p --legend-position=<pos> Specify the position of the legend (accepts any argument that the R function legend's position argument accepts). If you want a legend, this option is also required.
-v --save-legend Draws the legend to a separate file called <prefix>_legend.pdf (often useful so it doesn't overdraw your haplotype plot)
This option is more for debugging purposes, but it plots labels for each haplotype. These typically occur as Roman numerals.
option:
-h --haplotype-labels
haplotype.r produces three possible different output files.
Once you have finished rearranging your haplotype plot and right-click on the window, the plot is saved as a PDF with the name <prefix>_plot.pdf.
If you've chosen to plot a legend separately, you'll get another PDF called <prefix>_legend.pdf that contains your figure lenged by itself. You can then add this back into your haplotype plot in Illustrator or whatever.
haplotype.r will save the R data objects it used while calculating and plotting the haplotype network. The objects saved are:
- categories: a factor variable of the categories used to color haplotype pies
- hap: the haplotypes themselves
- hap.net: the haplotype network object
- hap.pies: an object that determines pie chart divisions/colors when plotting
- pal: the color palette (by default generated based on one of Beyoncé's outfits)
- plottr: the data object returned from
replot, which stores where all the haplotype pies are once you've rearranged them (so you don't have to rearrange them again if you want to plot it again).
Limitations (this section is now out of date, since I modified the pegas library to fix this issue, but I'm leaving it in for posterity)
There are a few things this script doesn't do as well as a couple of quirks it has.
- Unlike NETWORK, this script will not create circular networks
- It will not show missing/inferred haplotypes
- (This problem is obviated if you use the modified version of pegas available elsewhere on my github page) When drawing tickmarks on links for mutation steps, the script always draws the tickmark in the center of the link (as determined by the line between centers of the circles). This means that if you have a particularly large circle next to a small one, the tickmark may disappear inside the larger circle unless you drag the smaller one a fair distance away. In this example image, you can see some of the smaller haplotypes at their default distance with hidden tickmarks as well as some dragged far enough away for the tickmarks to be seen.