NEW UPDATE: VAB code added to newer version of CP2K-7.1.
Code addition to a cloned verison of CP2K-6.1 to calculate electron coupling matrix element VAB for molecular and periodic systems. [1] Calculation method is based on Farazdel & Dupuis work. [2]
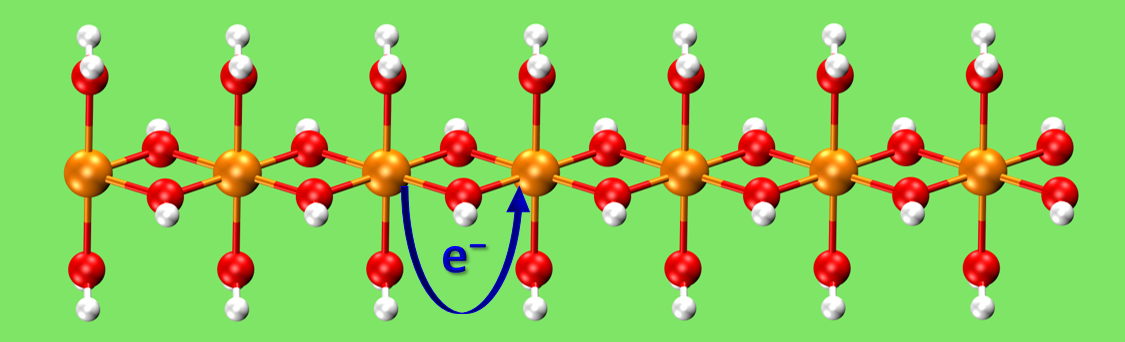
Testcases are in the folder ET_CP2K. Input section needs to be specified as follows:
Hartree Fock options are a replication of Guidon et al.'s HF section from CP2K manual https://manual.cp2k.org/cp2k-6_1-branch/CP2K_INPUT/FORCE_EVAL/DFT/XC/HF.html
&FORCE_EVAL
...
&MIXED
&VAB
DO_VAB .TRUE.
&HF !
FRACTION 1.0 !MUST BE 1.0 FOR VAB_CALCULATION
!Other optional sections for setting up HF calculation as in
!Guidon, et al.’s papers on HF implementation in CP2K 111,134,139
HF_INFO
INTERACTION_POTENTIAL
LOAD_BALANCE
MEMORY
PERIODIC
SCREENING
&END
&END
&END
...
&END
Maximal Orbital Analysis (MOA) for better interpretation of electronic interactions based on Dupuis et al.'s, work [3] is available.
Testcases are in the folder MOA_CP2K. Input section is as follows:
&PROPERTIES
&MOA
NFRG 2 ! No. of fragments
LFRG 1 2 ! Length of fragments
IFRG 1 2 3 ! Atoms in fragments by indices
NOMOA 1 1 1 0 0 ! Exclude MOs if index is non-zero
NOMOAA 1..3 0 0 ! Exclude alpha MOs if index is non-zero
NOMOAB 1..2 0 4..5 ! Exclude beta MOs if index is non-zero
IFUNO .FALSE. ! To do COT on alpha and beta within the system
REF_STATE_WFN_FILE <Filename> ! To use a reference state. COT will
! be done on respective alphas and betas
&MO_CUBES ! build wfn from MOA orbitals and print individual cubes
STRIDE 2 2 2 ! The stride (X,Y,Z) used to write the cube file
MO_LIST 1 2 3 ! List of MOs to print as cube
&END
&END MOA
&END PROPERTIES
To download the code
git clone https://github.com/dupuislab/CP2K.git
To build the code with the default Makefile and an arch file use
make -j8 ARCH=local VERSION=popt
where arch file local.popt exists in the arch folder.
For more detailed instructions on the dependencies and how to modify Makefiles to suit your compute nodes please visit the CP2K website
https://www.cp2k.org/howto
References:
- PK Behara, M Dupuis, Electron transfer in extended systems: characterization by periodic density functional theory including the electronic coupling, Phys. Chem. Chem. Phys., 2020, Advance Article.(https://dx.doi.org/10.1039/C9CP05133C)
- Abbas Farazdel, Michel Dupuis, Enrico Clementi, and Ari Aviram Journal of the American Chemical Society 1990 112 (11), 4206-4214 DOI: 10.1021/ja00167a016
- Michel Dupuis, Meghana Nallapu, Journal of Computational Chemistry 2019, 40, 39–50