q2 picrust2 Tutorial
The basic PICRUSt2 plugin for QIIME 2 is available here. This plugin can be integrated with the output files of the default QIIME 2 pipelines - i.e. with denoising approaches rather than closed-reference OTU picking. Installation instructions are available on the QIIME 2 plugin library website.
Before running this tutorial we recommend that you take a look through the standalone tutorial for a better description of the tool (click Tutorial on the right side-bar). Note that many of the features available in the standalone version are not implemented in the QIIME 2 plugin yet. You should also read through the key limitations of metagenome inference.
Running the full PICRUSt2 pipeline requires more computational requires than required by PICRUSt1, because the genome prediction steps are run for every dataset.
There are two pipelines for running PICRUSt2 in q2-picrust2:
-
A pipeline that involves running sequence placement with EPA-NG or SEPP using PICRUSt2 commands. The tutorial below will focus on running SEPP, which is less memory intensive.
-
A pipeline that involves running sequence placement with a tool outside of PICRUSt2 and then starting the pipeline at that point. You can see an example of how to do that (with an older version of QIIME 2) here.
You can see a description of the PICRUSt2 command by running:
qiime picrust2 full-pipeline --help
The required inputs are --i-table and --i-seq, which need to correspond to QIIME 2 artifacts of types FeatureTable[Frequency] and FeatureData[Sequence], respectively. The Feature Table needs to contain the abundances of ASVs (i.e. a BIOM table) and the sequence file needs to be a FASTA file containing the sequences for each ASV. There are many options available for the standalone PICRUSt2 version, but many options are missing from the QIIME 2 plugin.
-
--p-placement-toolindicates which placement tool (epa-ngorsepp) should be used for placing query sequences into reference tree. The default isepa-ng. -
--p-hsp-methodindicates the hidden-state prediction method to use. We will usepicin this tutorial, because it is fastest, but you may want to use the defaultmpon your own data. -
--p-max-nstispecifies how distantly placed a sequence needs to be in the reference phylogeny before it is excluded. The default cut-off is 2. In human datasets used for testing PICRUSt2 the only ASVs above this default cut-off were 18S sequences erroneously in 16S datasets, which suggests this cut-off is highly lenient. For environmental datasets a higher proportion of ASVs may be thrown out based on this default cut-off. -
--p-edge-exponentis a setting for maximum parsimony hidden-state prediction. Specifies weighting transition costs by the inverse length of edge lengths. If 0, then edge lengths do not influence predictions. Users running placement with SEPP and prediction with maximum parsimony may need to set this option to 0 to get around an error. -
--p-threadsindicates the number of threads (or CPUs) to use.
We will be using test files from the PICRUSt2 tutorial for this example. You can download these files using the below commands:
mkdir q2-picrust2_tutorial
cd q2-picrust2_tutorial
wget http://kronos.pharmacology.dal.ca/public_files/picrust/picrust2_tutorial_files/mammal_biom.qza
wget http://kronos.pharmacology.dal.ca/public_files/picrust/picrust2_tutorial_files/mammal_seqs.qza
wget http://kronos.pharmacology.dal.ca/public_files/picrust/picrust2_tutorial_files/mammal_metadata.tsv
These files correspond to the ASV count table, ASV sequences, and metadata for the samples. There are 11 samples and 371 ASVs in total. These samples were collected from the mammalian stool of Arctic wolves, coyotes, beavers, and porcupines, as previously described.
You can run the full PICRUSt2 pipeline with the below command (should take ~15 min and 3.8 GB of RAM - this will be much faster if you can set more threads).
qiime picrust2 full-pipeline \
--i-table mammal_biom.qza \
--i-seq mammal_seqs.qza \
--output-dir q2-picrust2_output \
--p-placement-tool sepp \
--p-threads 1 \
--p-hsp-method pic \
--p-max-nsti 2 \
--verbose
Note in this case the sepp placement method and the pic the phylogenetic independent contrast hidden-state prediction method are indicated because they are faster than the default (and SEPP is much less memory intensive). We recommend that in practice users use the mp method (note that setting the --p-edge-exponent to be 0 might be necessary for getting predictions based on a SEPP-placed tree). Also, if users have at least 16 GB of RAM they should place reads with EPA-NG.
The output artifacts of this command are the red boxes in the flowchart here.
These output files in (q2-picrust2_output) are:
-
ec_metagenome.qza- EC metagenome predictions (rows are EC numbers and columns are samples). -
ko_metagenome.qza- KO metagenome predictions (rows are KOs and columns are samples). -
pathway_abundance.qza- MetaCyc pathway abundance predictions (rows are pathways and columns are samples).
The artifacts are all of type FeatureTable[Frequency], which means they can be used with QIIME 2 plugins that process and analyze these datatypes.
For instance, you can get summary information regarding the pathway abundance file with this command, which you can view like any QIIME 2 visualization.
qiime feature-table summarize \
--i-table q2-picrust2_output/pathway_abundance.qza \
--o-visualization q2-picrust2_output/pathway_abundance.qzv
You should see a table like the one below, which describes the total pathway abundance per-sample (as well as other useful summaries). The below plots were created with the QIIME 2 2019.7 version of the plugin and based on using EPA-ng for placing the reads, so your plots will look a little different. Note that this isn't the final result (that would be the FeatureTable[Frequency] files created above) you would analyze and is instead just a way to get some basic descriptions of the data.

Note that this file is not in units of relative abundance (e.g. percent) and is instead the sum of the predicted functional abundance contributed by each ASV multiplied by the abundance (the number of input reads) of each ASV.
The above metagenome predictions can be integrated into a number of QIIME 2 analysis. For instance, you can quickly calculate diversity metrics based on these tables. The minimum sample pathway abundance found above was 226702 (at least for the version of the commands that the plot corresponded to), so we will rarify to this cut-off when calculating the core diversity metrics:
qiime diversity core-metrics \
--i-table q2-picrust2_output/pathway_abundance.qza \
--p-sampling-depth 226702 \
--m-metadata-file mammal_metadata.tsv \
--output-dir pathabun_core_metrics_out \
--p-n-jobs 1
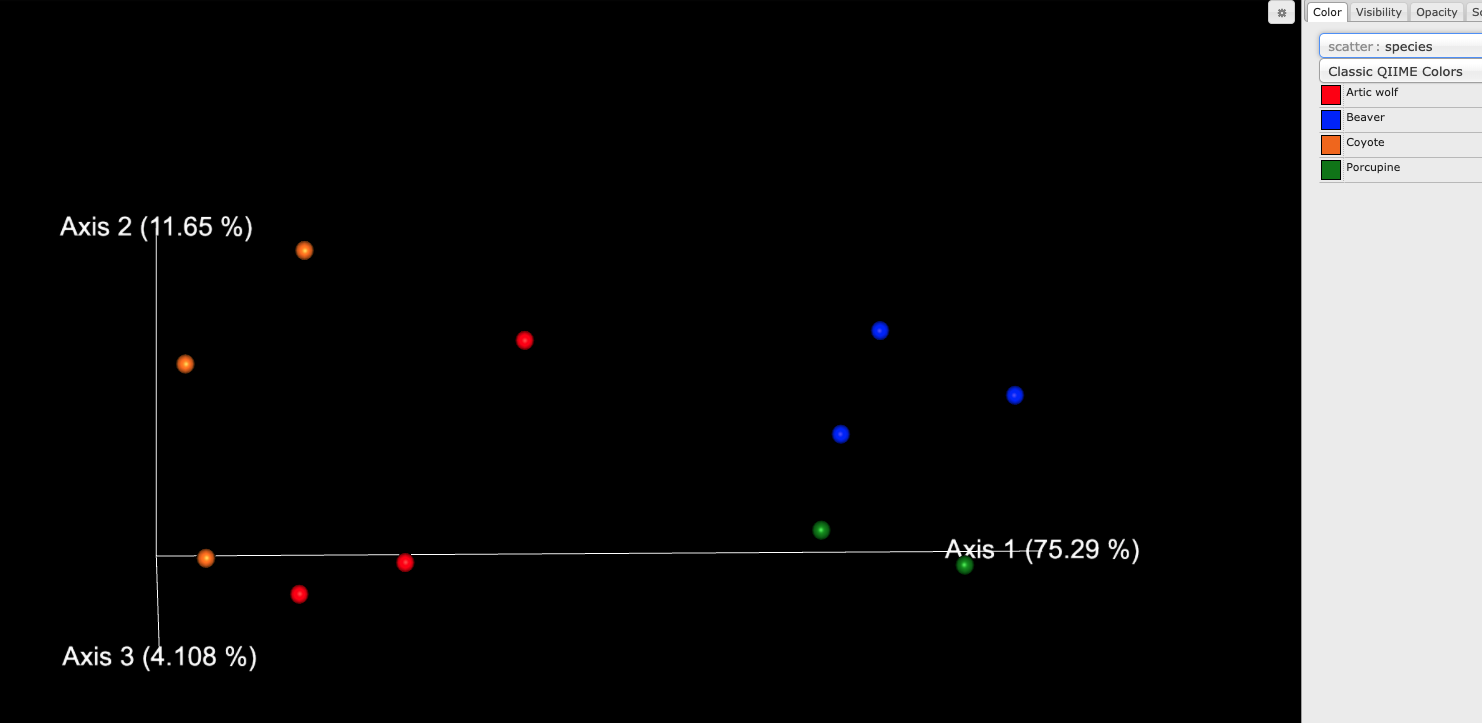
Take a look at pathabun_core_metrics_out/bray_curtis_emperor.qzv in the QIIME 2 viewer. You should see a ordination plot like this, which shows how the clear difference between this dataset is between carnivores and rodents:
The final output QZA files are QIIME 2 FeatureTable[Frequency] files, which can be used with existing QIIME 2 programs. Users are typically most interested in the predicted KEGG orthologs and MetaCyc pathways. If you want to use the tables outside of QIIME 2 you can convert the files to be BIOM format. For example, you can run this command to convert the pathway abundance table to BIOM format:
qiime tools export \
--input-path q2-picrust2_output/pathway_abundance.qza \
--output-path pathabun_exported
This command will convert a BIOM file to plain-text, which for the pathway abundance table would look like this:
biom convert \
-i pathabun_exported/feature-table.biom \
-o pathabun_exported/feature-table.biom.tsv \
--to-tsv
At this point many users are interested in identifying significantly different functions between sample groups. You should be aware that results based on differential abundance testing can vary substantially between shotgun metagenomics sequencing data and amplicon-based metagenome predictions based on the same samples. This is especially true for community-wide pathway predictions. Please check out this post and this response to a FAQ for more details.