{kind=link}
{kind=link}

When publishing results obtained with DFT-VASP WaNo, please consider citing it.
The DFT-VASP WaNo is a user-friendly tool that enables the performance of Density Functional Theory calculations using the widely-used VASP code without the need for a thorough understanding of VASP's functionalities and specifications. It offers a variety of methods and only requires the POSCAR file as mandatory input, with all other VASP input files being generated or loaded automatically. The commonly generated outputs include the OUTCAR, CONTCAR, CHGCAR, and POTCAR files and a lightweight, human-readable database in the yml extension, named vasp_results, that contains key information about the simulation.
Figure 1 illustrates the layout of the DFT-VASP WaNo, which features the INCAR, KPOINTS, Analysis, and Files-Run tabs. The INCAR tab allows the user to define the input parameters for the INCAR file and can be easily modified to accommodate additional input parameters by adding the necessary flags in the XML file. The remaining tabs, KPOINTS, Analysis, and Files-Run, enable the creation of the KPOINTS file and provide tools for performing DOS and Bader Charge Analysis, respectively. The Files-Run tab also controls the VASP compilation types (vasp_std, vasp_gam, and vasp_ncl) and allows the user to load the POSCAR file as an option, the INCAR, POTCAR, and KPOINTS files too.
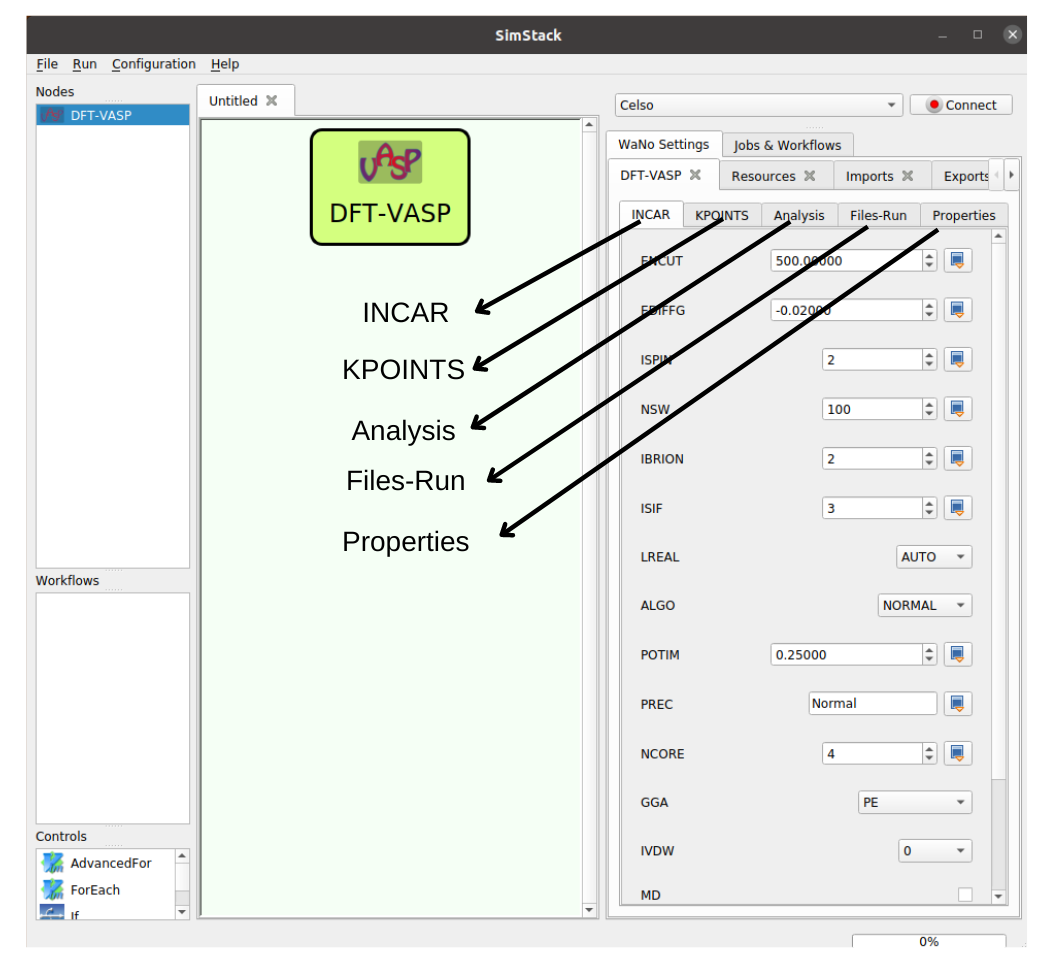
Fig 1 The DFT-VASP WaNo enables the performance of Density Functional Theory (DFT) calculations using the VASP code. It features an automated process for generating the POTCAR file after reading the POSCAR file. The user interface allows setting the KPOINTS and INCAR files and allows loading these input files in the Files-Run tab.
To get this WaNo up and running on your available computational resources, make sure to have the below libraries installed on Python 3.6 or newer.
1. Atomic Simulation Environment (ASE).
2. Python Materials Genomics (Pymatgen).
3. Numpy, os, sys, re, yaml.
- INCAR tab: See the GUI of this WaNo. as an option, we can set all INCAR flags available within VASP. However, we expose only a few of them, which are essential for the problem. A brief description of each flag pops up when we hover the mouse over the inputs.
- KPOINTS tab: Here the user can define two types of KPOINTS,
Kpoints_lengthandKpoints_Monkhorst. - Analysis tab: Aimed to compute Bader charge analysis and DOS.
- Files-Run tab: Mandatory loads the POSCAR file, and as an option can load INCAR, POTCAR, KPOINTS, and KORINGA files. The KORINGA file can be any file. In the case of this problem, it loads the Input_data.yml file.
- Properties tab: (:warning: The
vasp_results.ymldatabase is replacing this tab. Check down below to see the available properties in the database!). - All
.pyscripts may generate the INCAR, POTCAR, and KPOINTS files.
OUTCAR- This file contains vital information about the simulation of the system.
CONTCAR- The CONTCAR file contains information about the structure, e.g., the ionic positions.
POTCAR- The POTCAR file essentially contains the pseudopotential for each atomic species used in the calculation.
CHGCAR- VASP stores the charge density and the PAW one-center occupancies in the CHGCAR file, which can also be used to restart calculations.
vasp_results.yml- This database comprises all inputs in the
rendered_wano.ymlfile as well a set of keys and values of the following properties: total_energy, potential_energy, initial_magnetic_moments, magnetic_moment, magnetic_moments, kinetic_energy, cell, cell_lengths_and_angles, positions, forces, chemical_formula, chemical_symbols, center_of_mass, volume, temperature, all_distances, masses, atomic_numbers, global_number_of_atoms, initial_charges, band_gap, convergence, datetime, and user.
- This database comprises all inputs in the
- Step 1. Move the DFT-VASP folder to the WaNo directory.
- Step 2. Open Simstack on your compute and connect to your remote resource.
- Step 3. Drag the WaNo from the top left menu to the SimStack canvas as shown in Fig 1.
- Step 4. A double click on the WaNo will allow you to make the setups in the Input parameters.
- Step 5. Name your WaNo with
Ctrl+S, and running it withCtrl+Rcommand.
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 957189. The project is part of BATTERY 2030+, the large-scale European research initiative for inventing the sustainable batteries of the future.
Developer: Celso Ricardo C. Rêgo, Multiscale Materials Modelling and Virtual Design, Institute of Nanotechnology, Karlsruhe Institute of Technology https://www.int.kit.edu/wenzel.php
Licensed under the KIT License.