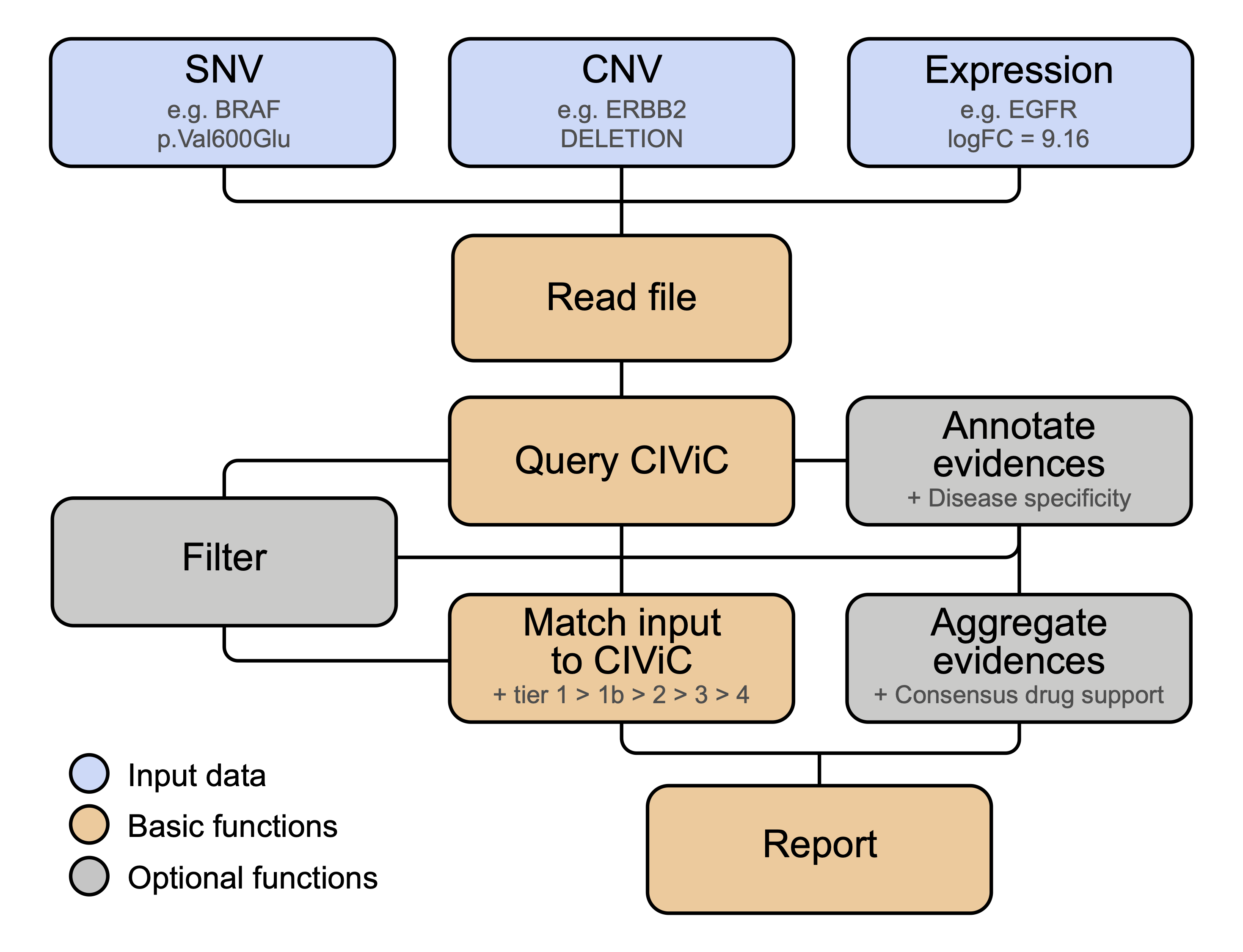
CIViCutils is a Python package for rapid retrieval, annotation, prioritization and downstream processing of information from the expert-curated CIViC knowledgebase (Clinical Interpretations of Variants in Cancer). CIViCutils can be integrated into novel and existing clinical workflows to provide variant-level disease-specific information about treatment response, pathogenesis, diagnosis, and prognosis of genomic aberrations (SNVs, InDels and CNVs), as well as differentially expressed genes. It streamlines interpreting large numbers of input alterations with querying and analyzing CIViC information, and enables the harmonization of input across different nomenclatures. Key features of CIViCutils include an automated matching framework for linking clinical evidence to input variants, as well as evaluating the accuracy of the resulting hits, and in-silico prediction of drug-target interactions tailored to individual patients and cancer subtypes of interest. For more details, see the CIViCutils publication.
- civicpy: To install, first activate the relevant Python (>=3.7) environment and then use pip install:
>> pip install civicpy
Then, to install CIViCutils, first activate the relevant Python (>=3.7) environment (i.e. already containing civicpy) and then use pip install:
>> pip install civicutils
The CIViC query implemented in CIViCutils makes use of an offline cache file of the CIViC database. The cache is provided by civicpy and retrieved with the initial installation of the CIViCutils package. Afterwards, users have to manually update the cache file if they want to leverage a new release version. To update the cache file, first activate the relevant Python environment, open a Python session, and then type:
from civicpy import civic
>> civic.update_cache()
More information can be found on the civicpy documentation.
Three different data types can be handled by the package: SNV (genomic single-nucleotide and insertion-deletion variants), CNV (genomic copy number alterations), and EXPR (differentially expressed genes). Corresponding functions for reading input data files are read_in_snvs(), read_in_cnvs() and read_in_expr(), respectively. Input files are required to have a tabular format with header and to contain data exclusively from one single data type. Example input files for all three data types are provided in subfolder data.
An input file of SNV/InDel data can be processed using CIViCutils function read_in_snvs(). Assumes header and the following columns:
Gene: required. One gene symbol per row allowed. Cannot be empty.Variant_dna: required. HGVS c. annotation for the variant (can be several possible annotations referring to the same variant, listed in a comma-separated list with no spaces). Can be empty, but at least one non-empty variant annotation must be provided acrossVariant_dnaandVariant_protper row.Variant_prot: required. HGVS p. annotation for the variant, if available (can can be several possible annotations referring to the same variant, listed in a comma-separated list with no spaces). Can be empty, but at least one non-empty variant annotation must be provided acrossVariant_dnaandVariant_protper row.Variant_impact: optional. Single or comma-separated list of variant impact annotations with no spaces. Such annotations (e.g.intron_variantorframeshift_variant) can be retrieved using tools like e.g. VEP or snpEff. Can be empty.Variant_exon: optional. Single or comma-separated list of variant exon annotations with no spaces. Such annotations (format:<N_EXON>/<TOTAL_EXONS>or<N_INTRON>/<TOTAL_INTRONS>, e.g.1/11) can be retrieved using tools like e.g. VEP or snpEff. If provided, thenVariant_impactmust exist and elements in both list must have a 1-1 correspondance. The reason is that the variant impact tag is used to determine if the exon annotation is intronic or exonic. Can be empty.
from civicutils.read_and_write import read_in_snvs
# Read-in file of input SNV variants
(raw_data, snv_data, extra_header) = read_in_snvs("data/example_snv.txt")
Function read_in_snvs() returns three elements: dictionary containing original rows and fields from input file (i.e. raw_data), dictionary of SNV/InDel data to be used for the CIViC query (i.e. snv_data), and list of additional columns provided in the input file which are not required for the CIViC query but should nonetheless be reported in case an output file is generated by CIViCutils (i.e. extra_header).
Structure of output dictionaries:
raw_data
└── <n_line>
└── [gene, variant_dna, variant_prot, variant_impact, variant_exon(, ...)] # as many appended fields as extra columns in the input file ('extra_header')
snv_data
└── <gene_id>
└── <variant_dna|variant_prot|variant_impact|variant_exon|n_line>
└── None
Note that variant_impact and variant_exon will be empty whenever these annotations were not provided by the user in the input file.
An input file of CNV data can be processed using CIViCutils function read_in_cnvs(). Assumes header and the following columns:
Gene: required. One gene symbol per row allowed. Cannot be empty.Variant_cnv: required. The following types of copy number variation annotations are allowed as input:AMPLIFICATION,AMP,GAIN,DUPLICATION,DUP,DELETION,DEL,LOSS. Several possible annotations referring to the same copy variant can be provided in a comma-separated list with no spaces. Cannot be empty.
from civicutils.read_and_write import read_in_cnvs
# Read-in file of input CNV variants
(raw_data, cnv_data, extra_header) = read_in_cnvs("data/example_cnv.txt")
Function read_in_cnvs() returns three elements: dictionary containing original rows and fields from input file (i.e. raw_data), dictionary of CNV data to be used for the CIViC query (i.e. cnv_data), and list of additional columns provided in the input file which are not required for the CIViC query but should nonetheless be reported in case an output file is generated by CIViCutils (i.e. extra_header).
Structure of output dictionaries:
raw_data
└── <n_line>
└── [gene, cnv(, ...)] # as many appended fields as extra columns in the input file ('extra_header')
cnv_data
└── <gene_id>
└── <cnv|n_line>
└── None
An input file of differential gene expression data can be processed using CIViCutils function read_in_expr(). Assumes header and the following columns:
Gene: required. One gene symbol per row allowed. Cannot be empty.logFC: required. Log fold-change value for the given gene. The sign of the fold-change is used to match variants in CIViC (eitherOVEREXPRESSIONif logFC>0 orUNDEREXPRESSIONif logFC<0). Cannot be empty and only one value allowed per row.
from civicutils.read_and_write import read_in_expr
# Read-in file of input differentially expressed genes
(raw_data, expr_data, extra_header) = read_in_expr("data/example_expr.txt")
Function read_in_expr() returns three elements: dictionary containing original rows and fields from input file (i.e. raw_data), dictionary of differential gene expression data to be used for the CIViC query (i.e. expr_data), and list of additional columns provided in the input file which are not required for the CIViC query but should nonetheless be reported in case an output file is generated by CIViCutils (i.e. extra_header).
Structure of output dictionaries:
raw_data
└── <n_line>
└── [gene, logfc(, ...)] # as many appended fields as extra columns in the input file ('extra_header')
expr_data
└── <gene_id>
└── <logfc|n_line>
└── None
CIViCutils leverages the offline cache file of the CIViC database provided by Python package civicpy, which allows performing high-throughput queries to the database. Information on how to install civicpy, as well as how to download and update the CIViC offline cache file, can be found above.
CIViCutils handles queries to CIViC through function query_civic(). Queries can only be gene-based and return all variants and associated clinical data which are annotated in the knowledgebase for each queried gene (only if any exist). Three types of identifiers are supported: entrez_symbol, entrez_id and civic_id. Note that the type of gene identifier initially chosen to perform the CIViC query must be selected throughout all the CIViCutils functions applied downstream.
from civicutils.query import query_civic
# Query a list of input genes in CIViC
var_map = query_civic(genes, identifier_type = "entrez_symbol")
# The gene list to be queried can be directly extracted from the output returned by CIViCutils' reading-in functions
var_map = query_civic(list(snv_data.keys()), identifier_type = "entrez_symbol")
Structure of the nested dictionary returned by the CIViC query (i.e. var_map):
var_map
└── <gene_id>
└── <var_id>
├── 'name' ── <var_name>
├── 'hgvs' ── [hgvs1, ..., hgvsN] # empty when no HGVS are available
├── 'types' ── [type1, ..., typeN] # 'NULL' when no types are available
└── <molecular_profile_id>
├── 'name' ── <molecular_profile_name>
├── 'civic_score' ── <molecular_profile_score>
├── 'n_evidence_items' ── <n_items>
└── 'evidence_items'
└── <evidence_type>
└── <disease> # can be 'NULL'
└── <drug> # 'NULL' when no drugs are available
└── <evidence> # <EVIDENCE_DIRECTION>:<CLINICAL_SIGNIFICANCE>
└── <level>
└── [evidence_item1, ...] # <PUBMED_ID>:<EVIDENCE_STATUS>:<SOURCE_STATUS>:<VARIANT_ORIGIN>:<RATING>
Query returns the following information for each variant retrieved from CIViC:
- Associated gene identifier
- CIViC variant identifier
- Name of the CIViC variant record
- CIViC Actionability Score: internal database metric computed across all evidence records for each variant to assess the quality and quantity of its associated clinical data
- Available HGVS expressions (can be empty when none are available)
- Available variant types: classification based on terms from the Sequence Ontology, e.g.
stop gained(can be empty when none are available) - Total number of evidence records associated with the variant
- Associated evidence records
In turn, the following information is returned by the query for each evidence record extracted from CIViC:
- Associated evidence type (either
Predictive,Diagnostic,Prognostic,Predisposing,Oncogenic, orFunctional) - Cancer indication: described using structured terms from the Disease Ontology database (can be empty for some evidence types)
- Drug/therapy (only available for
Predictiverecords, empty otherwise) - Clinical action, i.e. combination of evidence direction and clinical significance
- Evidence level
- Associated evidence items, i.e. individual publications used by curators to support the clinical claim (either PubMed identifiers or ASCO abstracts)
Last, in turn, the following information is returned by the query for each individual item:
- Evidence status: whether the given clinical statement has been submitted/unreviewed, rejected or accepted in the database
- Source status: whether the underlying source/publication is considered submitted, rejected or fully curated
- Variant origin: presumed origin of the alteration within the underlying study, e.g. inherited or acquired mutation
- CIViC confidence rating: score assigned by the curator summarizing the quality of the reported evidence in the knowledgebase
We refer to the CIViC documentation for detailed descriptions about the data contained in the knowledgebase.
CIViCutils enables flexible filtering of CIViC data based on several features via function filter_civic(). This offers users the possibility to clean-up and specifically select the set of CIViC records to be considered during the matching and annotation of variant-level data using CIViCutils. A comprehensive overview of available filtering features is provided below. Note that the supplied filtering parameters are evaluated in the order in which they are listed in the function definition, and not in the order specified during the function call. The logic for combining multiple filters is always AND; when the desired filtering logic in not possible in one single call, then the function needs to be applied to the data subsequently several times.
Complete list of filters available:
gene_id_inandgene_id_not_in: select or exclude specific gene identifiers (Entrez symbols, Entrez IDs or CIViC IDs), respectively.min_variants: select or exclude genes based on their number of associated CIViC variant records.var_id_inandvar_id_not_in: select or exclude specific CIViC variant identifiers, respectively.var_name_inandvar_name_not_in: select or exclude specific CIViC variant names, respectively.min_civic_score: select or exclude CIViC variants based on their associated CIViC score.var_type_inandvar_type_not_in: select or exclude CIViC variants based on their associated variant types, respectively.min_evidence_items: select or exclude CIViC variants based on their number of associated evidence records.evidence_type_inandevidence_type_not_in: select or exclude CIViC clinical records based on their associated evidence type, respectively.disease_inanddisease_not_in: select or exclude evidence records based on their associated cancer type, respectively.drug_name_inanddrug_name_not_in: select or exclude predictive records based on their associated drug name, respectively.evidence_dir_inandevidence_dir_not_in: select or exclude records based on their associated evidence direction, respectively.evidence_clinsig_inandevidence_clinsig_not_in: select or exclude evidence records based on their associated clinical significance, respectively.evidence_level_inandevidence_level_not_in: select or exclude records based on their associated evidence level, respectively.evidence_status_inandevidence_status_not_in: select or exclude records based on their associated evidence status, respectively.source_status_inandsource_status_not_in: select or exclude records based on the status of their supporting publication/source, respectively.var_origin_inandvar_origin_not_in: select or exclude CIViC records based on the presumed origin of the variant, respectively.source_type_inandsource_type_not_in: select or exclude records based on the types of supporting sources available, respectively.min_evidence_rating: select or exclude evidence records based on their associated rating.
from civicutils.filtering import filter_civic
filtered_map = filter_civic(var_map, evidence_status_in = ["ACCEPTED"], var_origin_in = ["SOMATIC"], output_empty=False)
Function filter_civic() additionally provides parameter output_empty, which indicates whether empty entries resulting from the applied filtering should be included in the dictionary returned by the function. Note that use of output_empty=True is not usually recommended, as other CIViCutils functions may behave unexpectedly or not work entirely when var_map containes empty entries. Instead, we recommend to only use this option for checking at which level the different records contained in var_map failed the applied filters.
CIViCutils provides function match_in_civic() to perform automated matching of the input genes and molecular alterations with variant-level evidence records retrieved from CIViC. Three different types of input data can be provided to the function: SNV, CNV and EXPR (see above for more information about the different data types and required formats of the input file in each case).
In order to link input and CIViC variants, the package attempts to standardize the names of the corresponding CIViC records by using a common nomenclature. To this end, the following are used: provided input HGVS expressions (only for data type SNV), HGVS expressions available in CIViC (if any exist, and only for data type SNV), and most importantly, a set of rules indicating how variants are normally named in the database records so that they can be appropiately translated into the format expected for the input alterations (e.g. input variant p.Val600Glu would correspond to CIViC record name V600E); for input data types CNV and EXPR, the matching to CIViC records is exclusively based on the most common variant names that exist in CIViC for each type of molecular aberration (e.g. OVEREXPRESSION, AMPLIFICATION, etc.).
CIViCutils uses a tier-based rating system to assess the quality of the resulting variant matches. The available tier categories are as follows (listed in descending hierarchical order):
tier_1: perfect match between input and CIViC variant(s), e.g.p.Val600Glumatched toV600E.tier_1b: a non-perfect match between input and CIViC variant(s), e.g. records likeMUTATION,FRAMESHIFT VARIANTorEXON 1 VARIANT.tier_2: positional match between input and CIViC variant(s), e.g.V600MandV600Kreturned whenV600Ewas provided. Note there is a special case of so-called "general" variants, e.g.V600, which are prioritized over any other positional hits which may have also been found by CIViCutils.tier_3: gene was found in CIViC but no associated variant record could be matched. In this case, all CIViC variant records available for the gene and found to match the given data type are returned by the function (if any). If atier_3was indicated but no matched variants are listed, then this is a consequence of no CIViC records being found for the provided data type and given gene (but indicates the existance of other CIViC records available for a different data type).tier_4: gene was not found in CIViC. No hits are returned by the query.
More details about the matching framework implemented in CIViCutils can be found here.
Note that the user can choose to perform filtering on the collected CIViC data before it is even supplied to match_in_civic() by providing a custom var_map that is used for the matching framework, e.g. if further filtering of the retrieved CIViC evidences needs to be applied so that undesired information is not considered downstream. We highly recommend this, specially to select only evidences tagged as ACCEPTED and avoid matching of submitted evidence that has not yet been expert-reviewed. In the case of genomic variants, it is also recommended to filter for the desired variant origin (e.g. SOMATIC, GERMLINE, etc.). Be aware that when filtering of CIViC evidence is performed prior to matching, then the returned matches and associated information might not reflect the exact state of the database, e.g. genes present in CIViC but specifically excluded from var_map by the user will be classified as tier 4. On the other hand, if var_map is not provided in the arguments of match_in_civic(), then per default the function directly retrieves from the database cache file all CIViC information available for the input genes, without applying any prior filtering.
from civicutils.match import match_in_civic
# Function automatically queries CIViC for the provided genes
(match_map, matched_ids, var_map) = match_in_civic(snv_data, data_type="SNV", identifier_type="entrez_symbol", select_tier="highest", var_map=None)
# Alternatively, the user can directly supply a custom set of CIViC evidences to match against using 'var_map'
(match_map, matched_ids, var_map) = match_in_civic(snv_data, data_type="SNV", identifier_type="entrez_symbol", select_tier="highest", var_map=var_map)
Structure of the nested dictionary returned by the variant matching framework (i.e. match_map):
match_map
└── <gene_id>
└── <input_variant>
└── <tier>
└── [var_id1, ...]
The returned dictionary (match_map) contains the same genes and variants provided in the input dictionary (i.e. either snv_data, cnv_data or expr_data, depending on the data type), with additional entries per gene and variant combination for every available tier category and listing the matches found in each case (if any).
CIViCutils offers functionality to filter and prioritize evidence records based on the corresponding tiers of their matched variants. Function match_in_civic() (performs the query to CIViC) allows the user to directly filter the returned variant matches based on their assigned tiers through option select_tier, which indicates the type of tier selection to be applied, and can be either: highest (returns the best tier reported per variant match, using established hierarchy 1>1b>2>3>4), all (does not apply any filtering and returns all tiers available for each variant match), or a list of specific tier categories to select for (if all are provided, then no filtering is done).
Alternatively, CIViCutils also provides function filter_matches(), which allows the user to select or filter variants based on their assigned tiers after the matching to CIViC evidence has already been performed, e.g. if match_in_civic() was initially run with argument select_tier=all, and now further filtering by tier is desired. This function offers the same filtering framework that can be applied during the CIViC query, i.e. parameter select_tier can be either highest, all or a list of specific tier categories to select for. Note that filter_matches() cannot be applied if the provided match_map was already processed and annotated for consensus drug response information.
from civicutils.match import filter_matches
# Filter based on the best assigned tier classification of the variant matches
filtered_map = filter_matches(match_map, select_tier = "highest")
# Alternatively, the user can filter variant matches deriving exclusively from specific tier classifications
# e.g. to remove gene-only variant matches and variants that could not be linked with CIViC data
filtered_map = filter_matches(match_map, select_tier = ["tier_1", "tier_1b", "tier_2"])
Variant-specific clinical data retrieved from CIViC can be further annotated with cancer type specificity information, based on their associated disease names and relative to one or more cancer indications of interest provided by the user. Excluding evidence records from undesired diseases can also be done at this step. To this end, the user can use function annotate_ct() and supply lists of non-allowed (disease_name_not_in), relevant (disease_name_in), and high-level/alternative (alt_disease_names) terms. More details about these parameters can be found below. As a result of the annotation, each available disease name retrieved from CIViC and the associated evidence are classified as either cancer type specific (ct), general specificity (gt) or non cancer type specific (nct).
The annotation of disease specificity using function annotate_ct() can only be applied if the provided var_map is not already annotated with this information. The function returns a similar nested dictionary with a slightly different structure, namely, containing one additional layer per evidence type which groups the disease names by their assigned category (see below).
from civicutils.match import annotate_ct
annotated_map = annotate_ct(var_map, disease_name_not_in, disease_name_in, alt_disease_names)
Structure of var_map after being annotated for disease specificity:
var_map
└── <gene_id>
└── <var_id>
├── 'name'
├── 'hgvs'
├── 'types'
└── <molecular_profile_id>
├── 'name'
├── 'civic_score'
├── 'n_evidence_items'
└── 'evidence_items'
└── <evidence_type>
└── <ct> # new layer included with the disease specificity label (ct, gt or nct)
└── <disease>
└── <drug>
└── <evidence>
└── <level>
└── <evidence_item>
In order to classify each disease and its associated evidences as cancer type specific (ct), general specificity (gt) or not cancer type specific (nct), lists of terms can be provided. Excluding evidences from undesired diseases can also be done at this step.
Relevant and non-allowed disease names or terms can be provided as lists in disease_name_in and disease_name_not_in, respectively. Relevant terms are used to find evidence records associated to specific cancer types and subtypes which might be of particular significance to the user. On the other hand, non-allowed terms are used to remove evidence records associated to undesired cancer types. In both cases, partial matches to the disease names in CIViC are sought, e.g. small will match non-small cell lung cancer and lung small cell carcinoma, while non-small will only match non-small cell lung cancer. In the same manner, be aware that uveal melanoma will only match uveal melanoma and not melanoma. As CIViC contains a small number of records associated to more general or high-level disease names, e.g. cancer or solid tumor, an additional list of alternative terms can be supplied to the package via alt_disease_names, which are used as a second-best classification when relevant cancer specificity terms cannot be found. Because these high-level disease names are database-specific, only exact matches are allowed in this case, cancer will only match cancer and not lung cancer. Input terms should always be provided in a comma-separated list, even if only one single term is supplied, and multiple words per term are permitted, e.g. [ovarian, solid tumor, sex cord-stromal] and [solid tumor] are both valid parameter inputs.
CIViC records are classified and selected/excluded based on cancer specificity according to the following logic, which is applied based on their associated disease name and the set of terms supplied by the user:
- If any non-allowed terms are provided in
disease_name_not_in, partial matches to the available disease names are sought, and any matched records are entirely excluded from the data (and hence from any downstream processing of CIViC information with CIViCutils). - For the remaining set of unclassified records, partial matches to the relevant terms in
disease_name_inare sought, and any matched records are classified and tagged as cancer type specific (ct). - For the remaining set of unclassified records, exact matches to the high-level terms in
alt_disease_namesare sought as a fall-back case, and any matched records are classified and tagged with general cancer specificity (gt). - All remaining evidence records which could not be classified are tagged as non-specific cancer type (
nct), regardless of the associated disease.
The above logic (hierarchy ct>gt>nct) is applied separately for each evidence type (i.e. Predictive, Diagnostic, Prognostic or Predisposing), which means that records of distinct evidence types can be associated to different sets of disease names, hence resulting in different cancer specificity classifications for the same variant.
To ease the selection of appropriate terms for classifying the disease specificity of a particular cancer type or subtype of interest, we provide a helper file civic_available_diseases_<DATE>.txt in the data subfolder of the TCGA-BLCA analysis, listing all disease names available in CIViC as of <DATE>. To update this file, run standalone script get_available_diseases_in_civic.py (which can be found in the scripts subfolder of the TCGA-BLCA analysis) as follows, replacing <DATE> with the new date:
> python tcga_analysis/scripts/get_available_diseases_in_civic.py --outfile tcga_analysis/data/civic_available_diseases_<DATE>.txt
Similarly as with the tier-based filtering, it is possible to select or exclude CIViC records based on their annotated cancer type specificity, e.g. to select only evidences from the best possible specificity per evidence type, or to focus on records associated with a particular cancer subtype. Once these annotations have been included into var_map using function annotate_ct(), the user can use function filter_ct() to filter or prioritize the available CIViC evidence based according to their assigned cancer type classifications. Parameter select_ct indicates the type of specificity selection to be performed on the supplied CIViC data, and can be either: highest (select only the evidences from the best available category per evidence type, using established hierarchy ct>gt>nct), all (do not apply any filtering and return all available disease classifications), or a list of specific categories to select for (if all are provided, then no filtering is done). Note that filtering is only possible if the provided var_map has been previously annotated with this information.
from civicutils.match import filter_ct
# Filter based on the best cancer type specificity found across CIViC data
filtered_map = filter_ct(var_map, select_ct = "highest")
# Alternatively, the user can filter CIViC data deriving exclusively from concrete specificity categories
# e.g. to remove evidence records classified as non cancer type specific based on the disease of interest
filtered_map = filter_ct(var_map, select_ct = ["ct", "gt"])
Available Predictive evidence retrieved from CIViC and matched to the input molecular alterations can be further processed and aggregated into so-called "consensus drug response predictions", which aim to effectively summarize the available drug information and facilitate in-silico prediction of candidate drugs and their therapeutic response tailored to specific variants and cancer types. To this end, CIViCutils provides function process_drug_support() which can combine multiple predictive evidence items into a single and unanimous response prediction for every tier match, drug, and disease specificity category available, using a majority vote scheme.
In addition, CIViCutils further interprets the evidence items to be aggregated (characterized by their combination of terms in the evidence direction and clinical significance) into a reduced set of concrete expressions relative to the direct therapeutic prediction; namely, POSITIVE, NEGATIVE, or UNKNOWN. To achieve this, the function makes use of a helper dictionary mapping CIViC evidence to drug responses, which is leveraged during the computation of the consensus predictions, and which can be customized by the user.
Structure and default values of drug_support entry in config file data.yml:
drug_support:
SUPPORTS:
SENSITIVITYRESPONSE: POSITIVE
RESISTANCE: NEGATIVE
REDUCED SENSITIVITY: NEGATIVE
ADVERSE RESPONSE: NEGATIVE
DOES_NOT_SUPPORT:
RESISTANCE: UNKNOWN_DNS
SENSITIVITYRESPONSE: UNKNOWN_DNS
REDUCED SENSITIVITY: UNKNOWN_DNS
ADVERSE RESPONSE: UNKNOWN_DNS
Be aware that CIViCutils can distinguish between two subtypes of UNKNOWN drug responses: those deriving from blank or null ("N/A") values for the evidence direction and/or clinical significance (UNKNOWN_BLANK), and optionally, those deriving from evidence direction DOES NOT SUPPORT (UNKNOWN_DNS, shown in the support dictionary above). Manual curation performed in Krentel et al. proved evidence direction DOES NOT SUPPORT to have an ambiguous meaning, dependant on the specific context of the underlying data, hence making it difficult to translate into a clearly defined consequence without the review of an expert. Nonetheless, CIViCutils allows the user to provide a different mapping of their choosing (however, the available categories to choose from are still restricted to either POSITIVE, NEGATIVE, UNKNOWN_BLANK, or UNKNOWN_DNS).
The consensus annotations resulting from the majority vote have the following format:
<DRUG>:<CT>:CIVIC_<CONSENSUS_PREDICTION>:<N_POSITIVE>:<N_NEGATIVE>:<N_UNKNOWN_BLANK>:<N_UNKNOWN_DNS>
where <DRUG> corresponds to the drug name or therapy retrieved from CIViC, <CT> to the corresponding cancer type specificity reported by CIViCutils (i.e. either CT, GT or NCT), and <CONSENSUS_PREDICTION> to the unanimous drug response assigned by the package based on the counts of evidence items available for each therapeutic prediction, which are also reported (<N_POSITIVE>, <N_NEGATIVE>, <N_UNKNOWN_BLANK> and <N_UNKNOWN_DNS>), resulting in the following response categories that can be reported as the final consensus prediction: SUPPORT (when most items are POSITIVE), RESISTANCE (when majority is NEGATIVE), CONFLICT (unresolved cases of confident and contradicting evidence) and UNKNOWN (prevailing category is UNKNOWN, i.e. aggregation of UNKNOWN_BLANK and UNKNOWN_DNS items, meaning that the predictive value is not known).
The annotation of consensus drug response predictions can only be performed if the provided var_map has been previously annotated with cancer type specificity information. The function returns a similar nested dictionary as match_in_civic(), but with a slightly different structure, namely, containing two additional layers per tier category: matches (containing the corresponding variant record hits found in CIViC, if any), and drug_support (listing one string for each consensus drug response generated for the given tier match) (see below).
from civicutils.read_and_write import get_dict_support
from civicutils.match import process_drug_support
# Get custom dictionary of support from data.yml (default already provided by CIViCutils)
# This defines how each combination of evidence direction + clinical significance in CIViC is classified in terms of drug support (e.g. sensitivity, resistance, unknown, etc.)
support_dict = get_dict_support()
# Process consensus drug response predictions for the matched variants based on the available CIViC evidence annotated with disease specificity
annotated_match = process_drug_support(match_map, var_map, support_dict)
Structure of match_map after being annotated for consensus drug response predictions (i.e. drug_support):
match_map
└── <gene_id>
└── <input_variant>
└── <tier>
├── 'matched' # new layer included to distinguish variant matches from drug information
│ └── [var_id1, ...]
└── 'drug_support' # new layer included with consensus drug response predictions
└── [response_prediction1, ...] # <DRUG>:<CT>:CIVIC_<CONSENSUS_PREDICTION>:<N_POSITIVE>:<N_NEGATIVE>:<N_UNKNOWN_BLANK>:<N_UNKNOWN_DNS>
The retrieved CIViC annotations can be written into a new output file with tabular format using function write_match(). The output table includes a header and uses a standardized structure which is identical regardless of the type of data at hand, including the same columns and contents of the input file originally supplied to the CIViCutils workflow, in addition to new columns which are appended by the package summarizing the data retrieved from the knowledgebase.
Required columns (dependent on the data type) are reported first in the output, while other columns that may have been present in the original input table can also be appended using parameter header (list is always retuned upon reading of the input data file). Subsequently, new CIViC-related columns are appended (see below), and in order to enable keeping track of the specific CIViC record from which each clinical statement in the output is derived, the reported entries include a prefix of the form <GENE>:<CIVIC_VARIANT> whenever applicable (namely, only not reported for columns CIViC_Tier and CIViC_Drug_Support). While <GENE> can take different values depending on the type of identifier selected by the user, the name of the retrieved variant record (i.e. <CIVIC_VARIANT>) remains unchanged regardless of the kind of queries performed to the knowledgebase.
from civicutils.read_and_write import write_match
write_match(match_map, var_map, raw_data, extra_header_cols, data_type="SNV", outfile, has_support=True, has_ct=True, write_ct=False, write_support=True, write_complete=False)
New columns appended by CIViCutils to the output file:
- CIViC_Tier: tier category assigned by CIViCutils for the listed variant match(es). Possible categories:
1,1b(only for data typeSNV),2(only data typesSNVandCNV),3or4. - CIViC_Score: semi-colon separated list of CIViC variant record(s) matched for the given input variant and their corresponding CIViC Actionability Scores.
- CIViC_VariantType: semi-colon separated list of variant types reported in CIViC for the matched variant record(s).
- CIViC_Drug_Support: semi-colon separated list of consensus drug response predictions generated by CIViCutils based on the available predictive CIViC evidence matched for the input variant. Optional column, only reported when
write_support = True. - CIViC_PREDICTIVE: semi-colon separated list of predictive evidence matched in CIViC for the input variant.
- CIViC_DIAGNOSTIC: semi-colon separated list of diagnostic evidence matched in CIViC for the input variant.
- CIViC_PROGNOSTIC: semi-colon separated list of prognostic evidence matched in CIViC for the input variant.
- CIViC_PREDISPOSING: semi-colon separated list of predisposing evidence matched in CIViC for the input variant.
The specific format used for the clinical statements listed in the evidence columns (i.e. CIViC_PREDICTIVE, CIViC_DIAGNOSTIC, CIViC_PROGNOSTIC, CIViC_PREDISPOSING) depends on the values supplied for parameters write_ct and write_complete in function write_match(). Furthermore, note that the format of evidences in column CIViC_PREDICTIVE deviates slightly from the format used in the other three columns (due to the existance of drugs associated with the evidence records in the former case).
- The evidence items reported in the CIViC evidence columns are aggregated whenever possible; namely, at the level of the disease name, combination of evidence direction and clinical significance, and evidence level. Format:
# For 'CIViC_DIAGNOSTIC', 'CIViC_PROGNOSTIC' and 'CIViC_PREDISPOSING' data
<DISEASE>(<DIRECTION>,<SIGNIFICANCE>(<LEVEL>(<SOURCE_ID>,...),...),...);
# For 'CIViC_PREDICTIVE' data (identical but including drug info in a new field)
<DISEASE>|<DRUG>(<DIRECTION>,<SIGNIFICANCE>(<LEVEL>(<SOURCE_ID>,...),...),...);
- The user can choose between the default "short" format (
write_complete=False) and a "long" format (write_complete=True) for reporting individual evidence items in the output table. In the first case, only the identifiers of the supporting sources/publications are listed (as shown above), while in the second, additional information is included for each item; namely, its evidence status, source status, variant origin and confidence rating. Format:
# Short format (default)
<SOURCE_ID>
# Long format
<SOURCE_ID>:<EVIDENCE_STATUS>:<SOURCE_STATUS>:<VARIANT_ORIGIN>:<RATING>
- Argument
write_ct=Truecan be selected to report the cancer type specificity label assigned to each disease (<CT>) in the items of the CIViC evidence columns. Note that when inputvar_mapis annotated with disease specificity, thenhas_ct=Truemust be selected inwrite_match()(and viceversa). Format:
# For 'CIViC_DIAGNOSTIC', 'CIViC_PROGNOSTIC' and 'CIViC_PREDISPOSING' data
<DISEASE>|<CT>(<DIRECTION>,<SIGNIFICANCE>(<LEVEL>(<SOURCE_ID>,...),...),...);
# For 'CIViC_PREDICTIVE' data (identical but including drug info in a new field)
<DISEASE>|<CT>|<DRUG>(<DIRECTION>,<SIGNIFICANCE>(<LEVEL>(<SOURCE_ID>,...),...),...);
- Argument
write_support=Truecan be selected to include one additional column in the output table, which lists the consensus drug response predictions computed by CIViCutils for each tier match (one prediction generated for each combination of available drug and disease specificity). When inputmatch_mapwas annotated with consensus drug response information, thenhas_support=Truemust be selected inwrite_match()(and viceversa). Format:
<DRUG>:<CT>:CIVIC_<CONSENSUS_PREDICTION>:<N_POSITIVE>:<N_NEGATIVE>:<N_UNKNOWN_BLANK>:<N_UNKNOWN_DNS>
write_to_json(): reports dictionary (e.g. of data retrieved from CIViC) into an output file using JSON format.write_to_yaml(): reports dictionary (e.g. of data retrieved from CIViC) into an output file using YAML format.
# Load package and import relevant functions
import civicutils
from civicutils.read_and_write import read_in_snvs, get_dict_support, write_match, write_drug_targets
from civicutils.query import query_civic
from civicutils.filtering import filter_civic
from civicutils.match import match_in_civic, annotate_ct, filter_ct, process_drug_support
# Read-in file of input SNV variants
(raw_data, snv_data, extra_header) = read_in_snvs("data/example_snv.txt")
# Query input genes in CIViC
var_map = query_civic(list(snv_data.keys()), identifier_type="entrez_symbol")
# Filter undesired evidences to avoid matching later on
var_map = filter_civic(var_map, evidence_status_in=["ACCEPTED"], var_origin_not_in=["GERMLINE"], output_empty=False)
# Match input SNV variants in CIViC, pick highest tier available per input gene+variant
# Tier hierarchy: 1 > 1b > 2 > 3 > 4
(match_map, matched_ids, var_map) = match_in_civic(snv_data, data_type="SNV", identifier_type="entrez_symbol", select_tier="highest", var_map=var_map)
# Annotate matched CIViC evidences with cancer specificity of the associated diseases
disease_name_not_in = []
disease_name_in = ["bladder"]
alt_disease_names = ["solid tumor"]
annot_map = annotate_ct(var_map, disease_name_not_in, disease_name_in, alt_disease_names)
# Filter CIViC evidences to pick only those for the highest cancer specificity available
# ct hierarchy: ct > gt > nct
annot_map = filter_ct(annot_map, select_ct="highest")
# Get custom dictionary of support from data.yml (provided within the package)
# This defines how each combination of evidence direction + clinical significance in CIViC is classified in terms of drug response (e.g. sensitivity, resistance, unknown, etc.)
support_dict = get_dict_support()
# Process consensus drug support for the matched variants using the underlying CIViC evidences annotated. report_drug_targets is a boolean value to indicate whether drugs targeting more than 2 gene variants should be reported.
(annot_match, drug_targets) = process_drug_support(match_map, annot_map, support_dict, report_drug_targets=True)
# Write to output
# Do not report the CT classification of each disease, and write column with the drug responses predicted for each available CT class of every variant match
write_match(annot_match, annot_map, raw_data, extra_header, data_type="SNV", outfile, has_support=True, has_ct=True, write_ct=False, write_support=True, write_complete=False)
# Create a secondary output file that compiles drugs targeting more than 2 gene variants. The file comprises variant details alongside the corresponding evidence linked to the Drug/Variant association.
write_drug_targets(drug_targets, raw_data, data_type="SNV", outfile_drug_targets)